Страница:
А. А. Дроздов,М. В. Дроздова
Органическая химия Шпаргалка
1. Биоорганическая химия
Это наука, изучающая биологическую функцию органических веществ в организме. Она возникла во второй половине XX в. Объектами ее изучения служат биополимеры, биорегуляторы и отдельные метаболиты.
Биополимеры – высокомолекулярные природные соединения, которые являются основой всех организмов. Это пептиды, белки, полисахариды, нуклеиновые кислоты (НК), липиды.
Биорегуляторы – соединения, которые химически регулируют обмен веществ. Это витамины, гормоны, антибиотики, алкалоиды, лекарственные препараты и др.
Знание строения и свойств биополимеров и биорегуляторов позволяет познать сущность биологических процессов. Так, установление строения белков и НК позволило развить представления о матричном биосинтезе белка и роли НК в сохранении и передаче генетической информации.
Основная задача биоорганической химии – выяснение взаимосвязи структуры и механизма действия соединений.
Это наука, изучающая соединения углерода. В настоящее время насчитывается – 16 млн органических веществ.
Причины многообразия органических веществ.
1. Соединения атомов углерода (С) друг с другом и другими элементами периодической системы Д. И. Менделеева. При этом образуются цепи и циклы.
2. Атом углерода может находиться в трех разных гибридных состояниях. Тетраэдрическая конфигурация атома С → плоскостная конфигурация атома С.
3. Гомология – это существование веществ с близкими свойствами, где каждый член гомологического ряда отличается от предыдущего на группу – СН2—.
4. Изомерия – это существование веществ, имеющих одинаковый качественный и количественный состав, но различное строение.
A. M. Бутлеров (1861 г.) создал теорию строения органических соединений, которая и по сей день служит научной основой органической химии. Основные положения теории строения органических соединений:
1) атомы в молекулах соединены друг с другом химическими связями в соответствии с их валентностью;
2) атомы в молекулах органических соединений соединяются между собой в определенной последовательности, что обусловливает химическое строение молекулы;
3) свойства органических соединений зависят не только от числа и природы входящих в их состав атомов, но и от химического строения молекул;
4) в молекулах существует взаимное влияние как связанных, так и непосредственно друг с другом не связанных атомов;
5) химическое строение вещества можно определить в результате изучения его химических превращений и, наоборот, по строению вещества можно охарактеризовать его свойства.
Биополимеры – высокомолекулярные природные соединения, которые являются основой всех организмов. Это пептиды, белки, полисахариды, нуклеиновые кислоты (НК), липиды.
Биорегуляторы – соединения, которые химически регулируют обмен веществ. Это витамины, гормоны, антибиотики, алкалоиды, лекарственные препараты и др.
Знание строения и свойств биополимеров и биорегуляторов позволяет познать сущность биологических процессов. Так, установление строения белков и НК позволило развить представления о матричном биосинтезе белка и роли НК в сохранении и передаче генетической информации.
Основная задача биоорганической химии – выяснение взаимосвязи структуры и механизма действия соединений.
Это наука, изучающая соединения углерода. В настоящее время насчитывается – 16 млн органических веществ.
Причины многообразия органических веществ.
1. Соединения атомов углерода (С) друг с другом и другими элементами периодической системы Д. И. Менделеева. При этом образуются цепи и циклы.
2. Атом углерода может находиться в трех разных гибридных состояниях. Тетраэдрическая конфигурация атома С → плоскостная конфигурация атома С.
3. Гомология – это существование веществ с близкими свойствами, где каждый член гомологического ряда отличается от предыдущего на группу – СН2—.
4. Изомерия – это существование веществ, имеющих одинаковый качественный и количественный состав, но различное строение.
A. M. Бутлеров (1861 г.) создал теорию строения органических соединений, которая и по сей день служит научной основой органической химии. Основные положения теории строения органических соединений:
1) атомы в молекулах соединены друг с другом химическими связями в соответствии с их валентностью;
2) атомы в молекулах органических соединений соединяются между собой в определенной последовательности, что обусловливает химическое строение молекулы;
3) свойства органических соединений зависят не только от числа и природы входящих в их состав атомов, но и от химического строения молекул;
4) в молекулах существует взаимное влияние как связанных, так и непосредственно друг с другом не связанных атомов;
5) химическое строение вещества можно определить в результате изучения его химических превращений и, наоборот, по строению вещества можно охарактеризовать его свойства.
2. Изомеры
Пространственные изомеры делятся на два вида: конформационные и конфигурационные.
1. Конформационными называются изомеры, формы молекул которых переходят друг в друга за счет свободного вращения атомов и групп атомов вокруг одной или нескольких б-связей. Первое соединение, для которого известно существование конформацион-ных изомеров, является этан. Его строение в пространстве изображается перспективной формулой или формулой Ньюмена.
2. Конфигурационные изомеры. Это стереоизо-меры, молекулы которых имеют различное расположение атомов в пространстве без учета конформаций.
Реоизомеры делятся на энантиомеры и диастерео-меры.
Энантномеры (оптические изомеры, зеркальные изомеры антиподы) – стереоизомеры, молекулы которых соотносятся между собой, как предмет и несовместимое с ним зеркальное отображение. Это явление называется энантиомерией.
Все химические и физические свойства энантиоме-ров одинаковы, кроме двух: вращение плоскости поляризованного света (в приборе поляриметре) и биологическая активность.
Условия энантиомерии:
1) атом С находится в состоянии sp3-гибридизации;
2) отсутствие всякой симметрии;
3) наличие асимметрического (хирального) атома С, атома, имеющего четыре разных заместителя.
Многие окси– и аминокислоты обладают способностью вращать плоскость поляризации луча света влево или вправо. Это явление называется оптической 2б активностью, а сами молекулы оптически активными. Отклонение луча света вправо отмечают знаком «+», влево – «-» и указывают угол вращения в градусах.
Абсолютную конфигурацию молекул определяют сложными физико-химическими методами.
Относительную конфигурацию оптически активных соединений определяют путем сравнения со стандартом глицеринового альдегида. Оптически активные вещества, имеющие конфигурацию правовращающего или левовращающего глицеринового альдегида (М. Розанов, 1906 г.), называется веществами D– и L-ряда. Равная смесь право– и левовращающих изомеров одного соединения называется рацематом и оптически неактивна.
Энантиомеры изображают с помощью формул Фишера. Среди энантиомеров могут быть симметричные молекулы, не обладающие оптической активностью, которые называются мезоизомерами. Оптические изомеры, не являющиеся зеркальными изомерами, отличающиеся конфигурацией нескольких, но не всех асимметрических атомов С, обладающие различными физическими и химическими свойствами, называется s-ди-а-стерео-изомерами.
p-диастереомеры (геометрические изомеры) – это стереомеры, имеющие в молекуле p-связь. Они встречаются в алкенах, непредельных высших карбоновых кислотах, непредельных дикарбоновых кислотах. Биологическая активность органических веществ связана с их строением.
1. Конформационными называются изомеры, формы молекул которых переходят друг в друга за счет свободного вращения атомов и групп атомов вокруг одной или нескольких б-связей. Первое соединение, для которого известно существование конформацион-ных изомеров, является этан. Его строение в пространстве изображается перспективной формулой или формулой Ньюмена.
2. Конфигурационные изомеры. Это стереоизо-меры, молекулы которых имеют различное расположение атомов в пространстве без учета конформаций.
Реоизомеры делятся на энантиомеры и диастерео-меры.
Энантномеры (оптические изомеры, зеркальные изомеры антиподы) – стереоизомеры, молекулы которых соотносятся между собой, как предмет и несовместимое с ним зеркальное отображение. Это явление называется энантиомерией.
Все химические и физические свойства энантиоме-ров одинаковы, кроме двух: вращение плоскости поляризованного света (в приборе поляриметре) и биологическая активность.
Условия энантиомерии:
1) атом С находится в состоянии sp3-гибридизации;
2) отсутствие всякой симметрии;
3) наличие асимметрического (хирального) атома С, атома, имеющего четыре разных заместителя.
Многие окси– и аминокислоты обладают способностью вращать плоскость поляризации луча света влево или вправо. Это явление называется оптической 2б активностью, а сами молекулы оптически активными. Отклонение луча света вправо отмечают знаком «+», влево – «-» и указывают угол вращения в градусах.
Абсолютную конфигурацию молекул определяют сложными физико-химическими методами.
Относительную конфигурацию оптически активных соединений определяют путем сравнения со стандартом глицеринового альдегида. Оптически активные вещества, имеющие конфигурацию правовращающего или левовращающего глицеринового альдегида (М. Розанов, 1906 г.), называется веществами D– и L-ряда. Равная смесь право– и левовращающих изомеров одного соединения называется рацематом и оптически неактивна.
Энантиомеры изображают с помощью формул Фишера. Среди энантиомеров могут быть симметричные молекулы, не обладающие оптической активностью, которые называются мезоизомерами. Оптические изомеры, не являющиеся зеркальными изомерами, отличающиеся конфигурацией нескольких, но не всех асимметрических атомов С, обладающие различными физическими и химическими свойствами, называется s-ди-а-стерео-изомерами.
p-диастереомеры (геометрические изомеры) – это стереомеры, имеющие в молекуле p-связь. Они встречаются в алкенах, непредельных высших карбоновых кислотах, непредельных дикарбоновых кислотах. Биологическая активность органических веществ связана с их строением.
3. Сопряженные системы
В простейшем случае сопряженные системы —
это системы с чередующимися двойными и одинарными связями. Они могут быть открытыми и закрытыми. Открытая система имеется в диеновых углеводородах (УВ).
Все атомы С находятся в состоянии sp-гибридиза-ции. Четыре негибридные р-орбитами, перекрываясь между собой, образуют единую электронную систему. Этот вид сопряжения называется p, p-сопряжением.
Происходит сопряжение р-электронов с S-электро-нами. Этот вид сопряжения называется р, р-сопряже-нием. Закрытая система имеется в ароматических УВ.
Сопряжение – процесс энергетически выгодный, энергия (Е) при этом выделяется. Энергия сопряжения бутадиена – 1,3 составляет 15 кДж/моль, энергия сопряжения бензола – 228 кДж/моль.
2. Ароматичность
Это понятие, включающее различные свойства ароматических соединений. Условия ароматичности:
1) плоский замкнутый цикл;
2) все атомы С находятся в sp2-гибридизации;
3) образуется единая сопряженная система всех атомов цикла;
4) выполняется правило Хюккеля: в сопряжении участвуют 4n + 2 р-электронов, где n = 1, 2, 3…
Простейший представитель ароматических углеводородов – бензол. Он соответствует всем четырем условиям ароматичности. Правило Хюккеля: 4n + 2 = 6, n = 1.
Нафталин – ароматическое соединение 4n + 2 = 10, n = 2.
Пиридин – ароматическое гетероциклическое соединение. Взаимное влияние атомов в молекуле
В 1861 г. русский ученый A. M. Бутлеров выдвинул положение: «Атомы в молекулах взаимно влияют друг на друга». В настоящее время это влияние передается двумя путями: индуктивным и мезомерным эффектами.
Индуктивный эффект – это передача электронного влияния по цепи р-связи. Известно, что связь между атомами с различной электроотрицательностью (ЭО) поляризована, смещена к более электроотрицательному атому. Это приводит к появлению на атомах эффективных (реальных) зарядов (d). Такое электронное смещение называется индуктивным и обозначается буквой «I» и стрелкой «→».
δ + δ –
СН3 – СН2 → X, Х = Hal-, НО-, HS-, NH2– и др.
Индуктивный эффект может быть положительным или отрицательным. Если заместитель X притягивает электроны химической связи сильнее, чем атом Н, то он проявляет – I.I (H) = 0. В нашем примере X проявляет – I.
Если заместитель X притягивает электроны связи слабее, чем атом Н, то он проявляет +I. Все алкилы (R = СН3-, C2H5– и т. д.), Меп+ проявляют +I.
это системы с чередующимися двойными и одинарными связями. Они могут быть открытыми и закрытыми. Открытая система имеется в диеновых углеводородах (УВ).
Все атомы С находятся в состоянии sp-гибридиза-ции. Четыре негибридные р-орбитами, перекрываясь между собой, образуют единую электронную систему. Этот вид сопряжения называется p, p-сопряжением.
Происходит сопряжение р-электронов с S-электро-нами. Этот вид сопряжения называется р, р-сопряже-нием. Закрытая система имеется в ароматических УВ.
Сопряжение – процесс энергетически выгодный, энергия (Е) при этом выделяется. Энергия сопряжения бутадиена – 1,3 составляет 15 кДж/моль, энергия сопряжения бензола – 228 кДж/моль.
2. Ароматичность
Это понятие, включающее различные свойства ароматических соединений. Условия ароматичности:
1) плоский замкнутый цикл;
2) все атомы С находятся в sp2-гибридизации;
3) образуется единая сопряженная система всех атомов цикла;
4) выполняется правило Хюккеля: в сопряжении участвуют 4n + 2 р-электронов, где n = 1, 2, 3…
Простейший представитель ароматических углеводородов – бензол. Он соответствует всем четырем условиям ароматичности. Правило Хюккеля: 4n + 2 = 6, n = 1.
Нафталин – ароматическое соединение 4n + 2 = 10, n = 2.
Пиридин – ароматическое гетероциклическое соединение. Взаимное влияние атомов в молекуле
В 1861 г. русский ученый A. M. Бутлеров выдвинул положение: «Атомы в молекулах взаимно влияют друг на друга». В настоящее время это влияние передается двумя путями: индуктивным и мезомерным эффектами.
Индуктивный эффект – это передача электронного влияния по цепи р-связи. Известно, что связь между атомами с различной электроотрицательностью (ЭО) поляризована, смещена к более электроотрицательному атому. Это приводит к появлению на атомах эффективных (реальных) зарядов (d). Такое электронное смещение называется индуктивным и обозначается буквой «I» и стрелкой «→».
δ + δ –
СН3 – СН2 → X, Х = Hal-, НО-, HS-, NH2– и др.
Индуктивный эффект может быть положительным или отрицательным. Если заместитель X притягивает электроны химической связи сильнее, чем атом Н, то он проявляет – I.I (H) = 0. В нашем примере X проявляет – I.
Если заместитель X притягивает электроны связи слабее, чем атом Н, то он проявляет +I. Все алкилы (R = СН3-, C2H5– и т. д.), Меп+ проявляют +I.
4. Мезомерный эффект
Мезомерный эффект (эффект сопряжения) – это влияние заместителя, передаваемое по сопряженной системе р-связей. Обозначается буквой ́«М» и изогнутой стрелкой. Мезомерный эффект может быть «+» или «-». Выше было сказано, что имеется два вида сопряжения р, р и р, р. Классификация органических реакций Химические реакции – это процессы, сопровождающиеся изменением распределения электронов внешних оболочек атомов реагирующих веществ. В результате реакции в реагирующих молекулах веществ разрываются одни химические связи и образуются другие. Реакция идет в сторону образования стабильных частиц, т. е. обладающих меньшей внутренней энергией.
Классифицировать реакции можно по различным признакам.
1. По типу разрыва химических связей в реагирующих частицах (субстрат и реагент). Субстрат – это реагирующее вещество, реагент – действующее вещество. Данное разделение условное.
Различают три типа реагентов:
1) радикалы (R) – это нейтральные атомы или частицы с неспаренным электроном (Н-, С1-.-ОН, – СН3 и др.);
2) нуклеофилы (Nu – «любящие ядра») – это частицы, имеющие электронную пару на внешнем электронном уровне атома;
3) электрофилы (Е – «любящие электроны») – это частицы, имеющие недостаток электронов – незаполненный валентный электронный уровень.
В реакциях нуклеофил атакует в субстрате реакционный центр с недостатком электронов, электрофил атакует реакционный центр с избытком электронов. Соответственно этому различают:
1) радикальные реакции;
2) электрофилъные реакции;
3) нуклеофильные реакции.
2. По количеству и характеру исходных и конечных продуктов различают типы реакций:
1) замещения; они подобны реакциям обмена в неорганической химии;
2) присоединения;
3) отщепления (элиминирования) – это отщепление двух атомов или групп атомов от соседних атомов углерода с образованием между ними р-связи;
4) перегруппировки.
С учетом характера реагентов реакции замещения и присоединения могут быть нуклеофильными, элект-рофильными и радикальными и обозначаться следующим образом:
1) реакции нуклеофильного замещения;
2) реакции электрофильного замещения;
3) реакции радикального замещения;
4) реакции электрофильного присоединения;
5) реакции нуклеофильного присоединения;
6) реакции оадикального присоединения.
Классифицировать реакции можно по различным признакам.
1. По типу разрыва химических связей в реагирующих частицах (субстрат и реагент). Субстрат – это реагирующее вещество, реагент – действующее вещество. Данное разделение условное.
Различают три типа реагентов:
1) радикалы (R) – это нейтральные атомы или частицы с неспаренным электроном (Н-, С1-.-ОН, – СН3 и др.);
2) нуклеофилы (Nu – «любящие ядра») – это частицы, имеющие электронную пару на внешнем электронном уровне атома;
3) электрофилы (Е – «любящие электроны») – это частицы, имеющие недостаток электронов – незаполненный валентный электронный уровень.
В реакциях нуклеофил атакует в субстрате реакционный центр с недостатком электронов, электрофил атакует реакционный центр с избытком электронов. Соответственно этому различают:
1) радикальные реакции;
2) электрофилъные реакции;
3) нуклеофильные реакции.
2. По количеству и характеру исходных и конечных продуктов различают типы реакций:
1) замещения; они подобны реакциям обмена в неорганической химии;
2) присоединения;
3) отщепления (элиминирования) – это отщепление двух атомов или групп атомов от соседних атомов углерода с образованием между ними р-связи;
4) перегруппировки.
С учетом характера реагентов реакции замещения и присоединения могут быть нуклеофильными, элект-рофильными и радикальными и обозначаться следующим образом:
1) реакции нуклеофильного замещения;
2) реакции электрофильного замещения;
3) реакции радикального замещения;
4) реакции электрофильного присоединения;
5) реакции нуклеофильного присоединения;
6) реакции оадикального присоединения.
5. Кислоты Бренстеда
Для характеристики кислотности и основности органических соединений применяют теорию Бренстеда.
Основные положения этой теории.
Кислота – это частица, отдающая протон (донор Н+); основание – это частица, принимающая протон (акцептор Н-).
Кислотность всегда характеризуется в присутствии оснований и наоборот.
А-Н(кислота) +В(основание) – А (сопряженное основание) + В-Н+ (сопряженная кислота).
Кислоты Бренстеда делятся на 4 вида в зависимости от кислотного центра:
1) SH-кислоты (тиолы);
2) ОН-кислоты (спирты, фенолы, карболовые кислоты);
3) НЗ-кислоты (амины, амиды);
4) Ф-СН-кислоты (УВ).
В этом ряду сверху вниз кислотность уменьшается. Сила кислоты определяется стабильностью образующегося аниона. Чем стабильнее анион, тем сильнее кислота. Стабильность аниона зависит от делока-лизации (распределения) «отрицательного» заряда по всей частице (аниону). Чем больше делокализован «отрицптельный» заряд, тем стабильнее анион и сильнее кислота.
Делокализация заряда зависит:
1) от электроотрицательности (ЭО) гетероатома. Чем больше ЭО гетероатома, тем сильнее соответствующая кислота. Например: R-OH и R-NH2.
Спирты более сильные кислоты, чем амины, т. к. ЭО (0) → 30(N);
2) от поляризуемости гетероатома. Чем больше поляризуемость гетероатома, тем сильнее соответствующая кислота. Например: R-SH и R-ОН.
Тиолы более сильные кислоты, чем спирты, т. к. атом S более поляризован, чем атом О;
3) от характера заместителя R (его длины, наличия сопряженной системы, делокализации электронной плотности).
Например: СН3-ОН, СН3-СН2-ОН, СН3-СН2-СН2-ОН. Кислотность меньше, так как увеличивается длина радикала.
При одинаковом кислотном центре сила спиртов, фенолов и карбоновых кислот не одинакова. Фенолы являются более сильными кислотами, чем спирты за счет р, s-сопряжения (+М) группы (-ОН). Связь О—Н более поляризуется в фенолах. Фенолы могут взаимодействовать даже с солями (FeC13) – качественная реакция на фенолы. Карбоновые кислоты по сравнению со спиртами, содержащими одинаковый R, являются более сильными кислотами, так как связь О—Н значительно поляризована за счет – М-эффекта группы > С = О. Кроме того, карбоксилат-анион более стабилен, чем анион спирта за счет р, s-сопряжения в карбоксильной группе;
4) от введения заместителей в радикал. ЭА-замести-тели увеличивают кислотность, ЭД-заместители уменьшают кислотность;
5) от характера растворителя.
Основные положения этой теории.
Кислота – это частица, отдающая протон (донор Н+); основание – это частица, принимающая протон (акцептор Н-).
Кислотность всегда характеризуется в присутствии оснований и наоборот.
А-Н(кислота) +В(основание) – А (сопряженное основание) + В-Н+ (сопряженная кислота).
Кислоты Бренстеда делятся на 4 вида в зависимости от кислотного центра:
1) SH-кислоты (тиолы);
2) ОН-кислоты (спирты, фенолы, карболовые кислоты);
3) НЗ-кислоты (амины, амиды);
4) Ф-СН-кислоты (УВ).
В этом ряду сверху вниз кислотность уменьшается. Сила кислоты определяется стабильностью образующегося аниона. Чем стабильнее анион, тем сильнее кислота. Стабильность аниона зависит от делока-лизации (распределения) «отрицательного» заряда по всей частице (аниону). Чем больше делокализован «отрицптельный» заряд, тем стабильнее анион и сильнее кислота.
Делокализация заряда зависит:
1) от электроотрицательности (ЭО) гетероатома. Чем больше ЭО гетероатома, тем сильнее соответствующая кислота. Например: R-OH и R-NH2.
Спирты более сильные кислоты, чем амины, т. к. ЭО (0) → 30(N);
2) от поляризуемости гетероатома. Чем больше поляризуемость гетероатома, тем сильнее соответствующая кислота. Например: R-SH и R-ОН.
Тиолы более сильные кислоты, чем спирты, т. к. атом S более поляризован, чем атом О;
3) от характера заместителя R (его длины, наличия сопряженной системы, делокализации электронной плотности).
Например: СН3-ОН, СН3-СН2-ОН, СН3-СН2-СН2-ОН. Кислотность меньше, так как увеличивается длина радикала.
При одинаковом кислотном центре сила спиртов, фенолов и карбоновых кислот не одинакова. Фенолы являются более сильными кислотами, чем спирты за счет р, s-сопряжения (+М) группы (-ОН). Связь О—Н более поляризуется в фенолах. Фенолы могут взаимодействовать даже с солями (FeC13) – качественная реакция на фенолы. Карбоновые кислоты по сравнению со спиртами, содержащими одинаковый R, являются более сильными кислотами, так как связь О—Н значительно поляризована за счет – М-эффекта группы > С = О. Кроме того, карбоксилат-анион более стабилен, чем анион спирта за счет р, s-сопряжения в карбоксильной группе;
4) от введения заместителей в радикал. ЭА-замести-тели увеличивают кислотность, ЭД-заместители уменьшают кислотность;
5) от характера растворителя.
6. Спирты
Спирты – это производные УВ, у которых один или несколько атомов Н замещено на – ОН-группу. Классификация.
1. По количеству групп ОН различают одноатомные, двухатомные и многоатомные спирты:
СН3 —СН2 —ОН (этанол);
СН2ОН-СН2ОН (этиленгликоль);
СН2ОН-СНОН-СН2ОН (глицерин).
2. По характеру R различают спирты: предельные, непредельные, циклические, ароматические.
3. По положению группы (-ОН) различают первичные, вторичные и третичные спирты.
4. По количеству атомов С различают низкомолекулярные и высокомолекулярные:
СН3(СН2)14-СН2-ОН либо (C16 H33OH);
цетиловый спирт
СН3-(СН2)29-СН2ОН (С31Н63, ОН).
мирициловый спирт
Цетилпальмитат – основа спермацета, мирицил-пальмитат – содержится в пчелином воске. Номенклатура
Тривиальный, рациональнаый, МН (корень + окончание «-ол» + арабская цифра). Изомерия
Возможны варианты: изомерии цепи, положения группы – ОН, оптическая изомерия.
Спирты – слабые кислоты.
Спирты – слабые основания. Присоединяют Н+ лишь от сильных кислот, но они более сильные Nu.
(—I) эффект группы (-ОН) увеличивает подвижность Н у соседнего углеродного атома. Углерод приобретает d+ (электрофильный центр, SE) и становится центром нуклеофильной атаки (Nu). Связь С-О рвется более легко, чем Н-О, поэтому характерными для спиртов являются реакции SN. Они, как правило, идут в кислой среде, так как протонирование атома кислорода увеличивает d+ атома углерода и облегчает разрыв связи. К этому типу относятся реакции образования эфиров, галогенопроизводных.
Смещение электронной плотности от Н в радикале приводит к появлению СН-кислотного центра. В этом случае идут реакции окисления и элиминирования.
Физические свойства
Низшие спирты (С1—С12) – жидкости, высшие – твердые вещества.
Химические свойства
Кислотно-основные.
Спирты – слабые амфотерные соединения.
Алкоголяты легко гидролизуются, это доказывает, что спирты более слабые кислоты, чем вода:
R-ONa + НОН → R—OH + NaOH.
1. По количеству групп ОН различают одноатомные, двухатомные и многоатомные спирты:
СН3 —СН2 —ОН (этанол);
СН2ОН-СН2ОН (этиленгликоль);
СН2ОН-СНОН-СН2ОН (глицерин).
2. По характеру R различают спирты: предельные, непредельные, циклические, ароматические.
3. По положению группы (-ОН) различают первичные, вторичные и третичные спирты.
4. По количеству атомов С различают низкомолекулярные и высокомолекулярные:
СН3(СН2)14-СН2-ОН либо (C16 H33OH);
цетиловый спирт
СН3-(СН2)29-СН2ОН (С31Н63, ОН).
мирициловый спирт
Цетилпальмитат – основа спермацета, мирицил-пальмитат – содержится в пчелином воске. Номенклатура
Тривиальный, рациональнаый, МН (корень + окончание «-ол» + арабская цифра). Изомерия
Возможны варианты: изомерии цепи, положения группы – ОН, оптическая изомерия.
Спирты – слабые кислоты.
Спирты – слабые основания. Присоединяют Н+ лишь от сильных кислот, но они более сильные Nu.
(—I) эффект группы (-ОН) увеличивает подвижность Н у соседнего углеродного атома. Углерод приобретает d+ (электрофильный центр, SE) и становится центром нуклеофильной атаки (Nu). Связь С-О рвется более легко, чем Н-О, поэтому характерными для спиртов являются реакции SN. Они, как правило, идут в кислой среде, так как протонирование атома кислорода увеличивает d+ атома углерода и облегчает разрыв связи. К этому типу относятся реакции образования эфиров, галогенопроизводных.
Смещение электронной плотности от Н в радикале приводит к появлению СН-кислотного центра. В этом случае идут реакции окисления и элиминирования.
Физические свойства
Низшие спирты (С1—С12) – жидкости, высшие – твердые вещества.
Химические свойства
Кислотно-основные.
Спирты – слабые амфотерные соединения.
Алкоголяты легко гидролизуются, это доказывает, что спирты более слабые кислоты, чем вода:
R-ONa + НОН → R—OH + NaOH.
7. Химические свойства спиртов
Группа – ОН является «плохо уходя щей группой» (связь малополярна), поэтому большинство реакций проводят в кислой среде.
Механизм реакции:
СН3 —СН2 —ОН+ Н+ → СН3 —СН2 + Н2O.
карбокатион
Если реакция идет с галогеноводородами, то присоединяться будет галогенид-ион: СН3 —СН2 + Сl → СН3 —СН2 СI1.
Анионы в таких реакциях выступают в качестве нуклеофилов (Nu) за счет «-» заряда или неподеленной электронной пары. Анионы являются более сильными основаниями и нуклеофильными реагентами, чем сами спирты. Поэтому на практике для получения простых и сложных эфиров используются алкоголяты, а не сами спирты. Если нуклеофилом является другая молекула спирта, то она присоединяется к карбокатиону:
СН3 —СН2 + R-0– Н → CH3 —CH2 —O-R.
простой эфир
Реакции Е (отщепления, или элиминирования). Эти реакции конкурируют с реакциями SN.
СН3 —СН2 —ОН + Н+ → СН3 —СН2 —O – Н → СН3 —СН2 + Н2O.
Реакция протекает при повышенной температуре и катализаторе H2SO4.
При избытке H2SO4 и более высокой температуре, чем в случае реакции образования простых эфиров, идет регенерация катализатора и образуется алкен:
СН3 —СН2 + HS04 → СН2 = СН2 + H2SO4.
Легче идет реакция Е для третичных спиртов, труднее – для вторичных и первичных, т. к. в последних случаях образуются менее стабильные катионы. В данных реакциях выполняется правило А. М. Зайцева: «При дегидратации спиртов атом Н отщепляется от соседнего атома С с меньшим содержанием атомов Н».
В организме группа – ОН под действием фермента превращается в легкоуходящую путем образования эфиров с Н3РО4.
СН3-СН2-ОН + НО-РО3Н2 → СН3-СН2-ОРО3Н2.
Реакции окисления:
1. Первичные и вторичные спирты окисляются СиО, растворами KMnO4К2Сr2O7 при нагревании с образованием соответствующих карбонилсодержащих соединений.
СН3 —СН2 —СН2 —ОН + О → СН3 —СН2 —НС = О + Н2О;
СН3—HСOН—СН3 + О → СН3—СO—СН3 + Н2О.
2. Третичные спирты окисляются с трудом.
К реакциям окисления относятся и реакции дегидрирования.
СН3 —СН2 —ОН ־ СН3 → НС = О + Н2.
IV. По радикалу (R) протекают реакции, характерные для соответствующих углеводородов (УВ).
СН3-СН2-ОН + 3Br2 → СВr3-СН2-ОН + ЗНВг;
СН2 = СН-СН2-ОН + Вr2– → СН2Вг-СНВг-СН2ОН.
Механизм реакции:
СН3 —СН2 —ОН+ Н+ → СН3 —СН2 + Н2O.
карбокатион
Если реакция идет с галогеноводородами, то присоединяться будет галогенид-ион: СН3 —СН2 + Сl → СН3 —СН2 СI1.
Анионы в таких реакциях выступают в качестве нуклеофилов (Nu) за счет «-» заряда или неподеленной электронной пары. Анионы являются более сильными основаниями и нуклеофильными реагентами, чем сами спирты. Поэтому на практике для получения простых и сложных эфиров используются алкоголяты, а не сами спирты. Если нуклеофилом является другая молекула спирта, то она присоединяется к карбокатиону:
СН3 —СН2 + R-0– Н → CH3 —CH2 —O-R.
простой эфир
Реакции Е (отщепления, или элиминирования). Эти реакции конкурируют с реакциями SN.
СН3 —СН2 —ОН + Н+ → СН3 —СН2 —O – Н → СН3 —СН2 + Н2O.
Реакция протекает при повышенной температуре и катализаторе H2SO4.
При избытке H2SO4 и более высокой температуре, чем в случае реакции образования простых эфиров, идет регенерация катализатора и образуется алкен:
СН3 —СН2 + HS04 → СН2 = СН2 + H2SO4.
Легче идет реакция Е для третичных спиртов, труднее – для вторичных и первичных, т. к. в последних случаях образуются менее стабильные катионы. В данных реакциях выполняется правило А. М. Зайцева: «При дегидратации спиртов атом Н отщепляется от соседнего атома С с меньшим содержанием атомов Н».
В организме группа – ОН под действием фермента превращается в легкоуходящую путем образования эфиров с Н3РО4.
СН3-СН2-ОН + НО-РО3Н2 → СН3-СН2-ОРО3Н2.
Реакции окисления:
1. Первичные и вторичные спирты окисляются СиО, растворами KMnO4К2Сr2O7 при нагревании с образованием соответствующих карбонилсодержащих соединений.
СН3 —СН2 —СН2 —ОН + О → СН3 —СН2 —НС = О + Н2О;
СН3—HСOН—СН3 + О → СН3—СO—СН3 + Н2О.
2. Третичные спирты окисляются с трудом.
К реакциям окисления относятся и реакции дегидрирования.
СН3 —СН2 —ОН ־ СН3 → НС = О + Н2.
IV. По радикалу (R) протекают реакции, характерные для соответствующих углеводородов (УВ).
СН3-СН2-ОН + 3Br2 → СВr3-СН2-ОН + ЗНВг;
СН2 = СН-СН2-ОН + Вr2– → СН2Вг-СНВг-СН2ОН.
8. Многоатомные спирты
Для этих спиртов характерны все реакции одноатомных спиртов, однако имеется ряд особенностей.
За счет (-I) группы (-ОН) многоатомные спирты обладают более выраженными кислотными свойствами.
Они образуют алкоголяты не только со щелочными металлами, но и со щелочами:
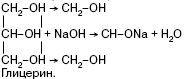
Качественной реакцией на двухатомные и многоатомные спирты (диольный фрагмент) является реакция с Си(ОН)2 в щелочной среде, в результате которой образуется комплексное соединение гликолят меди в растворе, дающем синее окрашивание.
Реакции многоатомных спиртов могут протекать по одной или всем группам (-ОН). Они образуют алкоголяты, простые и сложные эфиры, дегидратируются, окисляются.
Нитроглицерин – бесцветная маслянистая жидкость. В виде разбавленных спиртовых растворов (1 %-ных) применяется при стенокардии, так как оказывает сосудорасширяющее действие. Нитроглицерин – сильное взрывчатое вещество, способное взрываться от удара или при нагревании. При этом в малом объеме, который занимает жидкое вещество, мгновенно образуется очень большой объем газов, что и вызывает сильную взрывную волну. Нитроглицерин входит в состав динамита, пороха.
Представители пентитов и гекситов – ксилит и сорбит – соответственно, пяти– и шестиатомные спирты с открытой цепью. Накопление (-ОН) – групп ведет к появлению сладкого вкуса. Ксилит и сорбит – заменители сахара для больных диабетом.
Глицерофосфаты – структурные фрагменты фос-фолипидов, применяются как общеукрепляющее средство. В результате действия Н3 РО4 на глицерин получают смесь глицерофосфатов. Глицерофосфаты
Глицерофосфат железа (III) применяется при анемии, астении, общем упадке сил. Детям по 0,3–0,5 г 2–3 раза в день, взрослым по 1 г 3–4 раза.
Глицерофосфат кальция – при переутомлении, рахите, упадке питания. Детям по 0,05—0,2 г на прием, взрослым по 0,2–0,5.
1. При действии на глицерин KHSО4 и при нагревании – образуется акролеин.
2. При окислении глицерина образуется ряд продуктов. При мягком окислении – глицериновый альдегид и дигидроксиацетон. При окислении в жестких условиях образуется 1,3-диоксоацетон.
За счет (-I) группы (-ОН) многоатомные спирты обладают более выраженными кислотными свойствами.
Они образуют алкоголяты не только со щелочными металлами, но и со щелочами:
Качественной реакцией на двухатомные и многоатомные спирты (диольный фрагмент) является реакция с Си(ОН)2 в щелочной среде, в результате которой образуется комплексное соединение гликолят меди в растворе, дающем синее окрашивание.
Реакции многоатомных спиртов могут протекать по одной или всем группам (-ОН). Они образуют алкоголяты, простые и сложные эфиры, дегидратируются, окисляются.
Нитроглицерин – бесцветная маслянистая жидкость. В виде разбавленных спиртовых растворов (1 %-ных) применяется при стенокардии, так как оказывает сосудорасширяющее действие. Нитроглицерин – сильное взрывчатое вещество, способное взрываться от удара или при нагревании. При этом в малом объеме, который занимает жидкое вещество, мгновенно образуется очень большой объем газов, что и вызывает сильную взрывную волну. Нитроглицерин входит в состав динамита, пороха.
Представители пентитов и гекситов – ксилит и сорбит – соответственно, пяти– и шестиатомные спирты с открытой цепью. Накопление (-ОН) – групп ведет к появлению сладкого вкуса. Ксилит и сорбит – заменители сахара для больных диабетом.
Глицерофосфаты – структурные фрагменты фос-фолипидов, применяются как общеукрепляющее средство. В результате действия Н3 РО4 на глицерин получают смесь глицерофосфатов. Глицерофосфаты
Глицерофосфат железа (III) применяется при анемии, астении, общем упадке сил. Детям по 0,3–0,5 г 2–3 раза в день, взрослым по 1 г 3–4 раза.
Глицерофосфат кальция – при переутомлении, рахите, упадке питания. Детям по 0,05—0,2 г на прием, взрослым по 0,2–0,5.
1. При действии на глицерин KHSО4 и при нагревании – образуется акролеин.
2. При окислении глицерина образуется ряд продуктов. При мягком окислении – глицериновый альдегид и дигидроксиацетон. При окислении в жестких условиях образуется 1,3-диоксоацетон.
9. Предельные (насыщенные) углеводороды
Простейший представитель подгруппы предельных углеводородов – метан (СН4). И3 метана можно получить все другие предельные углеводороды, и в связи с этим все предельные углеводороды часто называются углеводородами ряда метана.
Для получения из метана других углеводородов вначале на метан нужно воздействовать хлором. При этом атом водорода в метане заменяется атомом хлора и получается хлористый метил.
Если теперь подействовать на полученный хлористый метил металлическим натрием, то натрий отнимет хлор, и образующиеся группы СН3, так называемые метальные радикалы, будут соединяться попарно одна с другой за счет освободившихся валентностей.
Химической стойкостью предельных углеводородов к ряду сильных реагентов, таких как крепкие кислоты и щелочи, относят парафины (от лат. parum affinis – «мало сродства»). При реакции получится предельный углеводород с двумя атомами углерода – этан (С2Н6).
Если, действуя на этан хлором, получим хлористый этил C2H5Сl1 а затем, смешав его с хлористым метилом, отнимем хлор натрием, то получим следующего представителя предельных углеводородов, содержащего три атома углерода, – пропан С3Н8.
Как видно из приведенных примеров, обе реакции сводятся в конечном итоге к замене в исходном углеводороде атома водорода метильной группой. Подобным образом в две стадии можно получить и последующие представители предельных углеводородов: бутан С4Н10 , пентан С5Н12.
Эти углеводороды представляют собой так называемый гомологический ряд. В таком ряду каждое последующее соединение можно получить из предыдущего путем одних и тех же химических реакций. Все соединения гомологического ряда, кроме того, близки по своим свойствам. Формула каждого соединения отличается от формулы предыдущего на одну и ту же группу атомов СН2, которая называется гомологической разностью. Соединения, являющиеся членами гомологического ряда, называются гомологами. Номенклатура и изомерия
Желая показать сходство всех предельных углеводородов с их родоначальником метаном, этим углеводородам дали названия, оканчивающиеся на – ан. Что касается начальной части наименований, то они возникли различным путем. Наименования первых трех гомологов метана – этана (С2Н6), пропана (С3Н8) и бутана (С4Н10) – возникли более или менее случайно. Начиная с С5Н12, названия углеводородов происходят от греческих (или в некоторых случаях латинских) названий чисел, соответствующих числу атомов углерода в данном соединении. Так, углеводород с пятью атомами углерода называется пентан (от греч. пента – пять); углеводород с шестью атомами углерода называется гексан (от греч. гекса – «шесть»); углеводород с семью атомами углерода называется гептан (от греч. гепта – «семь») и т. д.
При отнятии от углеводородов одного атома водорода получаются остатки предельных углеводородов, называемые одновалентными радикалами, или иногда просто радикалами.
Для получения из метана других углеводородов вначале на метан нужно воздействовать хлором. При этом атом водорода в метане заменяется атомом хлора и получается хлористый метил.
Если теперь подействовать на полученный хлористый метил металлическим натрием, то натрий отнимет хлор, и образующиеся группы СН3, так называемые метальные радикалы, будут соединяться попарно одна с другой за счет освободившихся валентностей.
Химической стойкостью предельных углеводородов к ряду сильных реагентов, таких как крепкие кислоты и щелочи, относят парафины (от лат. parum affinis – «мало сродства»). При реакции получится предельный углеводород с двумя атомами углерода – этан (С2Н6).
Если, действуя на этан хлором, получим хлористый этил C2H5Сl1 а затем, смешав его с хлористым метилом, отнимем хлор натрием, то получим следующего представителя предельных углеводородов, содержащего три атома углерода, – пропан С3Н8.
Как видно из приведенных примеров, обе реакции сводятся в конечном итоге к замене в исходном углеводороде атома водорода метильной группой. Подобным образом в две стадии можно получить и последующие представители предельных углеводородов: бутан С4Н10 , пентан С5Н12.
Эти углеводороды представляют собой так называемый гомологический ряд. В таком ряду каждое последующее соединение можно получить из предыдущего путем одних и тех же химических реакций. Все соединения гомологического ряда, кроме того, близки по своим свойствам. Формула каждого соединения отличается от формулы предыдущего на одну и ту же группу атомов СН2, которая называется гомологической разностью. Соединения, являющиеся членами гомологического ряда, называются гомологами. Номенклатура и изомерия
Желая показать сходство всех предельных углеводородов с их родоначальником метаном, этим углеводородам дали названия, оканчивающиеся на – ан. Что касается начальной части наименований, то они возникли различным путем. Наименования первых трех гомологов метана – этана (С2Н6), пропана (С3Н8) и бутана (С4Н10) – возникли более или менее случайно. Начиная с С5Н12, названия углеводородов происходят от греческих (или в некоторых случаях латинских) названий чисел, соответствующих числу атомов углерода в данном соединении. Так, углеводород с пятью атомами углерода называется пентан (от греч. пента – пять); углеводород с шестью атомами углерода называется гексан (от греч. гекса – «шесть»); углеводород с семью атомами углерода называется гептан (от греч. гепта – «семь») и т. д.
При отнятии от углеводородов одного атома водорода получаются остатки предельных углеводородов, называемые одновалентными радикалами, или иногда просто радикалами.
10. Национальная и международная номенклатура
Еще в середине XIX в. отдельные химики пытались создать такую номенклатуру, которая говорила бы о строении называемых веществ; такую номенклатуру называют рациональной. При этом, например, названия углеводородов производились от названий первого представителя данной группы углеводородов. Так, для ряда метана основой наименования служило название метана. Например, один из изомеров пентана можно назвать диметилэтилметан, т. е. это вещество можно представить как производное метана, у которого два атома водорода замещены метальными группами СН3, а один атом водорода – этиль-ной группой С2Н5.
Международная номенклатура
Желая создать наиболее рациональную номенклатуру органических соединений, которая была бы принята во всех странах мира, крупнейшие химики – представители химических обществ разных стран – собрались в 1892 г. в Женеве (Швейцария). На этом совещании была выработана систематическая научная номенклатура, которую теперь обычно называют женевской или международной номенклатурой.
Для того чтобы назвать какое-либо соединение по женевской номенклатуре, руководствуются следующими правилами.
Рассматривая структурную формулу соединения, выбирают наиболее длинную цепь атомов углерода и нумеруют атомы, начиная с того конца, к которому ближе стоит заместитель (боковое ответвление).
Соединение рассматривается согласно принципам женевской номенклатуры как производное нормального углеводорода, имеющего такую же, соответствующую перенумерованной цепь.
Место заместителя (ответвления цепи) обозначают цифрой, соответствующей номеру атома углерода, у которого стоит заместитель, затем называют заместитель и, наконец, углеводород, от которого производят все соединение по наиболее длинной перенумерованной цепи.
В тех случаях, когда в цепи имеется несколько ответвлений, положение каждого указывается отдельно соответствующими цифрами, и каждый заместитель называется особо. Если соединение имеет несколько одинаковых заместителей, например две метильные группы, то после двух цифр, обозначающих их места, говорят «диметил» (от греч. ди – «два»); при наличии трех метильных групп говорят «три-метил» и т. д.
Международная номенклатура
Желая создать наиболее рациональную номенклатуру органических соединений, которая была бы принята во всех странах мира, крупнейшие химики – представители химических обществ разных стран – собрались в 1892 г. в Женеве (Швейцария). На этом совещании была выработана систематическая научная номенклатура, которую теперь обычно называют женевской или международной номенклатурой.
Для того чтобы назвать какое-либо соединение по женевской номенклатуре, руководствуются следующими правилами.
Рассматривая структурную формулу соединения, выбирают наиболее длинную цепь атомов углерода и нумеруют атомы, начиная с того конца, к которому ближе стоит заместитель (боковое ответвление).
Соединение рассматривается согласно принципам женевской номенклатуры как производное нормального углеводорода, имеющего такую же, соответствующую перенумерованной цепь.
Место заместителя (ответвления цепи) обозначают цифрой, соответствующей номеру атома углерода, у которого стоит заместитель, затем называют заместитель и, наконец, углеводород, от которого производят все соединение по наиболее длинной перенумерованной цепи.
В тех случаях, когда в цепи имеется несколько ответвлений, положение каждого указывается отдельно соответствующими цифрами, и каждый заместитель называется особо. Если соединение имеет несколько одинаковых заместителей, например две метильные группы, то после двух цифр, обозначающих их места, говорят «диметил» (от греч. ди – «два»); при наличии трех метильных групп говорят «три-метил» и т. д.