Страница:
Приводим рисунок 2 (схема наследственной передачи семейной ГХС), который показывает путь передачи гетерозиготной доминантной мутации (лица мужского пола обозначены квадратами, лица женского пола – кружками). Носители мутантного гена семейной ГХС на этом рисунке имеют двуцветную характеристику.
В семьях, где один из родителей имеет доминантный мутантный ген (вызывающий ГХС), а второй родитель обладает здоровым набором генов, вероятность наследования патологического доминантного гена, как упоминалось, составляет 50 %. Иначе говоря, у половины потомков в таком браке можно ожидать рождения «гетерозигот» с семейной ГХС.
Итак, первичные ДЛП могут быть обусловлены одним «главным» мутантным геном (моногенные семейные ДЛП), причем мутации этого гена могут быть разными, т. е. они гетерогенны.
В большинстве случаев первичные ДЛП являются следствием взаимодействия нескольких нуклеотидных полиморфизмов в ансамбле второстепенных полигенов, участвующих в регуляции липидного метаболизма. Это полигенные ДЛП в семьях с наследственным предрасположением к ДЛП, к которому присоединяются неблагоприятные факторы внешней среды (например, избыток животных жиров и ХС в рационе и пр.) или отягощающие сопутствующие заболевания (метаболический синдром, сахарный диабет, ожирение).
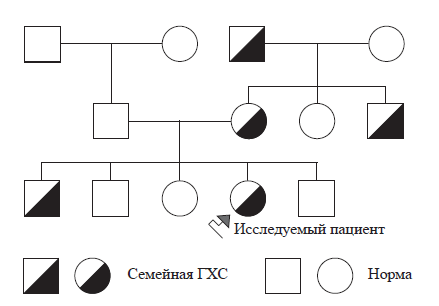
Рис. 2. Схема наследственной передачи семейной ГХС
Если появление ДЛП при моногенном семейном наследовании неизбежно, то развитие ДЛП при наследовании нуклеотидных полиморфизмов наступает необязательно.
1.4.1. Моногенные (семейные) ДАП
1.4.2. Полигенные ДЛП
1.4.3. Клинические проявления дислипидемий
В семьях, где один из родителей имеет доминантный мутантный ген (вызывающий ГХС), а второй родитель обладает здоровым набором генов, вероятность наследования патологического доминантного гена, как упоминалось, составляет 50 %. Иначе говоря, у половины потомков в таком браке можно ожидать рождения «гетерозигот» с семейной ГХС.
Итак, первичные ДЛП могут быть обусловлены одним «главным» мутантным геном (моногенные семейные ДЛП), причем мутации этого гена могут быть разными, т. е. они гетерогенны.
В большинстве случаев первичные ДЛП являются следствием взаимодействия нескольких нуклеотидных полиморфизмов в ансамбле второстепенных полигенов, участвующих в регуляции липидного метаболизма. Это полигенные ДЛП в семьях с наследственным предрасположением к ДЛП, к которому присоединяются неблагоприятные факторы внешней среды (например, избыток животных жиров и ХС в рационе и пр.) или отягощающие сопутствующие заболевания (метаболический синдром, сахарный диабет, ожирение).
Рис. 2. Схема наследственной передачи семейной ГХС
Если появление ДЛП при моногенном семейном наследовании неизбежно, то развитие ДЛП при наследовании нуклеотидных полиморфизмов наступает необязательно.
1.4.1. Моногенные (семейные) ДАП
Рассмотрим вначале конкретные типы моногенных ДЛП.
Для наших широт наиболее типична семейная ГХС по причине дефицита или дефекта рецепторов ЛПНП в печеночных и других соматических клетках или их низкой функциональной активности (в связи с дефектом структуры). Следствие этого – накопление ЛПНП-частиц – основных носителей ХС – в циркулирующей крови.
При относительно частых гетерозиготных формах семейной ГХС (1 случай на 500 человек населения) уровень ХС крови у этих лиц обычно колеблется в пределах 8-10 ммоль/л (350–500 мг/дл). В редких случаях гомозиготной семейной ГХС (1 случай на 1 млн жителей) уровень ХС крови достигает 15–20 ммоль/л (650-1000 мг/дл).
Нуклеотидная цепь гена рецептора ЛПНП состоит примерно из 45 тысяч нуклеотидов. Надо полагать, что при ее построении вероятность «ошибки» или мутации довольно велика, причем эти «сбои» могут возникать в разных участках нуклеотидной цепи, способны «закрепляться» и наследоваться. Описано около 1500 вариантов мутаций, ведущих к семейной ГХС и вызывающих нарушение работы рецепторов ЛПНП.
Один из вариантов этой мутации довольно широко распространен среди восточно-европейских евреев – «ашкенази», предки которых были выходцами из Литвы, из-за чего мутация получила название «литовской». Интересно, что среди евреев испанского происхождения – давних жителей Средиземноморья (евреев-«сефардов») – «литовская» мутация не встречается.
Среди франко-канадцев и жителей ЮАР с голландскими корнями частота этого заболевания очень высока (1 случай на 100–300 человек), что получило название «эффекта основателя». Это объясняют тем, что в относительно ограниченной популяции заключаются в основном только «внутренние» браки, т. е. эта этническая группа почти не смешивается с более широкими слоями населения, что увеличивает шанс появления генетически обусловленной патологии.
Мутации семейной ГХС, связанные с дефектом гена рецепторов ЛПНП, возникают в одном из генетических локусов 19-й хромосомы.
Другой вариант семейной ГХС характеризуется нарушением структуры апопротеина В-100 – ключевого белка ЛПНП. В этом случае нормальные рецепторы ЛПНП плохо распознают ЛПНП и плохо связываются с ними, поскольку апо В-100 является лигандом (связующим звеном) рецепторов ЛПНП. Ген, контролирующий структуру апо В-100, локализуется во 2-й хромосоме. Мутация этого гена влечет за собой дефект апо В-100, что приведет к плохой эвакуации ЛПНП из циркулирующей крови. Это другой вариант семейной ГХС. Данный тип мутации чаще всего встречается среди народов, населяющих страны Западной Европы.
Оба описанных варианта семейной ГХС имеют характер доминантных мутаций.
Описан третий (редкий) вид семейной ГХС, который носит аутосомно-рецессивный характер (ARH). Мутация была обнаружена в одной ливанской семье и получила название «ливанской». Мутация локализуется в одном из генов 1-й хромосомы. Этот ген «отвечает» за синтез транспортного белка, переносящего комплекс – рецептор ЛПНП + ЛПНП – с поверхности печеночных клеток внутрь, к органоидам, которые должны утилизировать ЛПНП. Если указанный транспортный белок не способен переносить комплексы рецептор ЛПНП+ ЛПНП внутрь клетки, они скапливаются на мембране и блокируют дальнейшее связывание ЛПНП. Следствием этого будет задержка ЛПНП в периферической крови, т. е. ГХС.
Недавно открыт еще один – 4-й – вид моногенной аутосомно-доминантной мутации, тоже приводящей к семейной ГХС. Это результат мутации одного из генов 1-й хромосомы, контролирующего синтез фермента «пропротеин-конвертазы субтилизин-кексинового типа 9», обозначаемого как PCSK-9. В зависимости от особенностей своей структуры этот фермент может либо тормозить, либо стимулировать активность рецепторов ЛПНП. В том случае, когда продуцируется PCSK-9 с повышенным сродством к рецепторам ЛПНП, деятельность этих рецепторов будет угнетаться, что приведет к ГХС.
Интересно, что мутация этого фермента может оказаться и прямо противоположной той, что описана выше. В результате этого сродство PCSK-9 к рецепторам ЛПНП понизится, они (рецепторы ЛПНП) будут весьма активно работать, что вызовет снижение уровня ЛПНП и ХС крови. В этом случае можно говорить о мутации с положительным знаком, которая обеспечит устойчивость к развитию атеросклероза.
Известна также моногенная мутация, связанная не с ГЛП, а с дислипидемией (ДЛП) без ГХС, приводящая к распространенному атеросклерозу из-за очень низкого содержания в плазме крови антиатерогенной фракции – ЛПВП. Это редкое эндемичное заболевание – Танжерская болезнь, которая распространена на одном из островов в Западной Атлантике. Заболевание развивается вследствие гомозиготной мутации гена, локализованного в 9-й хромосоме и кодирующего синтез транспортного белка АВСА-1. Этот белок выполняет функцию специализированного внутриклеточного переносчика свободного ХС к ключевому апопротеину ЛПВП – апо А1. Если в организме возникает дефицит АВСА, блокируется образование зрелых, функционально активных форм ЛПВП. Помимо нехватки транспортного белка (АВСА-1), при Танжерской болезни значительно снижается также содержание апобелков А-1 и A-II – ключевых белков ЛПВП, без которых их синтез невозможен.
Дефицит ЛПВП-3 и ЛПВП-2 резко снижает обратный транспорт ХС с поверхности периферических клеток в печень. ХС задерживается в тканях, в том числе накапливается в сосудистой стенке, что ведет к образованию атеросклеротических бляшек, хотя ГХС при этом не развивается.
В литературе описаны и другие гомозиготные мутации, связанные с дефектами других белковых транспортеров, переносящих стерины, – это ABCG-5, ABCG-8. При нарушенной структуре этих белков выход стеринов из клеток резко затрудняется [К. Berge et al., 2000].
Как правило, такие дефекты сопровождаются повышенным всасыванием растительных стеринов [J. Horton et al., 2002], что приводит к сито-стеролемии, хотя ГХС отсутствует. Поскольку необходимых белковых переносчиков стеринов мало, в организме нарушается способность выделять стерины, которые постепенно накапливаются в печени. Это влечет за собой подавление синтеза рецепторов ЛПНП, что в дальнейшем тоже может вызвать ГХС.
Дислипидемические состояния, характеризующиеся избирательным снижением уровня ЛПВП (без ГХС и без ГТГ) встречаются довольно часто во всех регионах мира, хотя они не связаны с Танжерской болезнью. При гетерозиготном носительстве дефектного гена АВСА-1 Танжерская болезнь не развивается, но содержание ЛПВП-фракции бывает отчетливо сниженным, что делает таких людей подверженными атеросклерозу [М.Ю. Мандельштам, В. Б. Васильев, 2008]. Даже один патологический аллель гена, управляющего синтезом белка АВСА-1, может оказать отрицательное влияние на этот транспортный белок и сделать его недостаточно функциональным.
В некоторых случаях дефицит ЛПВП развивается как результат аномалии в строении генов, контролирующих синтез лецитин-холестерин-ацилтрансферазы (ЛХАТ), участие которой необходимо для эстерификации свободного ХС. Этот дефект возникает как результат полигенной патологии при комбинации нескольких функционально значимых нуклеотидных полиморфизмов (в нескольких генетических локусах).
Еще один моногенный (генетически обусловленный) рецессивный дефект – причина редкой формы дислипидемии – абеталипопротеинемии. При этом типе ДЛП нарушается синтез апопротеина В, резко тормозится образование ЛПНП-частиц и снижается уровень ХС и ТГ крови. Хотя в этом случае исчезает почва для развития атеросклероза, развиваются серьезные нарушения со стороны центральной и периферической нервной системы, поскольку при крайнем дефиците ЛПНП в первую очередь страдает трофика нервных клеток и их отростков.
Из других мало известных моногенных дефектов, связанных с развитием ДЛП, можно указать на мутацию гена, управляющего продукцией скевенджер-рецепторов. Функция этих клеточных образований печени заключается в том, чтобы захватывать возвращающиеся в печень ЛПВП, нагруженные ХС, который они принесли с периферии. Если функция скевенджер-рецепторов нарушена, ЛПВП плохо проникают в печень, их концентрация в периферической крови повышается, но функция парализуется, т. к. они нагружены ХС до предела и больше не могут выполнять свою антиатерогенную миссию [D. Osgood et al., 2003]. Уровень общего ХС крови при этом повышается. Таким образом, иногда приходится сталкиваться с больными, у которых ЛПВП много, но функционально они неактивны. Иначе говоря, встречаются индивиды с высоким содержанием ЛПВП-частиц, а их защищенность от атеросклероза находится на очень низком уровне.
Похожая ситуация возникает при генетически обусловленной недостаточности триглицеридлипазы, которая плохо расщепляет ТГ, не освобождает от них ЛПВП, что резко тормозит антиатерогенную функцию этих частиц [R. Hegele et al., 1991].
Плохая работоспособность ЛПВП может быть связана не только с их дефицитом, но с генетически обусловленной патологией (недостатком) переносящих липиды транспортных белков. Так, при недостатке такого транспортного белка, как СЕТР (переносит ХС-эстеры и ТГ), затрудняется мобилизация эстерифицированного ХС из ЛПВП, эти частицы укрупняются, перегружаются ХС и вместо акцепторов ХС становятся его донорами [ILLUMINATE, 2006]. При этом концентрация ХС ЛПВП в периферической крови доходит до 70 мг/дл (1,8 ммоль/л) и выше. На этом примере еще раз можно убедиться в том, что не всегда высокое содержание антиатерогенной фракции липопротеидов (ЛПВП) в циркулирующей крови гарантирует защиту от атеросклероза. Одного такого больного мы наблюдали.
Приводим наше наблюдение № 1.
Больной С., 65 лет (1995 г.). В марте 1995 г. больной перенес острый инфаркт миокарда на переднебоковой стенке левого желудочка. При осмотре в домашних условиях через 3 дня после сердечного приступа (больной отказался от госпитализации) состояние больного удовлетворительное, пульс – 60 уд/мин, АД= 140/80 мм рт. ст. В анамнезе – много лет АГ, нерегулярно принимал гипотензивные препараты. В 1980 г. (в возрасте 50 лет) перенес первый инфаркт миокарда. В мае 1995 г. при исследовании липидного состава крови общий ХС = 308 мг/дл (7,9 ммоль/л), ТТ=185 мг/дл (2,1 ммоль/л), ХС ЛПВП = 91 мг/дл (2,3 ммоль/л), ХС ЛПНП= 180 мг/дл (4,6 ммоль/л), коэффициент атерогенности – 2,4 ед.
РЕЗЮМЕ. Несмотря на очень высокий уровень ХС ЛПВП (91 мг/дл) и нормальный коэффициент атерогенности, больной дважды перенес инфаркт миокарда. Есть все основания предполагать, что ЛПВП-частицы у данного больного не выполняют предназначенную им роль. Этих частиц много, но функционально они, по-видимому, неполноценны.
Встречаются и другие формы генетически детерминированных дислипидемий, например, комбинированная гиперлипидемия, при которой повышен уровень и ХС, и ТГ. Этот вид ДЛП (ГЛП) возникает в результате мутации гена, контролирующего синтез транскрипционной ДНК, которая описана под названием Upstream Stimulatory Factor (USF-1). Это также моногенная мутация, при которой главным образом нарушается элиминация ЛПОНП, а значит и ТГ, из циркулирующей крови. Впервые данная мутация была идентифицирована в Финляндии и в своем гетерозиготном варианте, по-видимому, обусловливает ГТГ у лиц с метаболическим синдромом. Авторы, описавшие эту мутацию [P. Pajukanta et al., 2004], рассматривают USF-1 как регулятор работы других генов, который посредством РНК кодирует синтез ряда белков – участников обмена и транспортировки апопротеинов А-2, С-3, Е, АВСА-1 и др.
Особого внимания требует оценка генетической детерминации апопротеина Е. По данным исследователей из Южной Кореи [Н. Oh et al., 2008], установлено несколько изоформ апоЕ (апо Е-2, апо Е-3, апо Е-4), которые могут встречаться в разных комбинациях, что зависит от наследственно обусловленного генотипа. Было установлено, что у большинства практически здоровых лиц, не имеющих ДЛП, обычно встречается генотип Е-З/Е-З (гомозиготный) или аллели Е-З/Е-2 либо Е-З/Е-4 (гетерозиготные). В некоторых случаях (из-за наличия определенных нуклеотидных полиморфизмов в каких-то генах) образуются генотипы Е-2/Е-2 или Е4/Е4, при которых продуцируются неполноценные апопротеины Е [D. Betteridge & J. Morrell, 2003]. В части таких случаев возникает предрасположенность к развитию тяжелой комбинированной гиперлипидемии, близкой к III типу по классификации Фредриксона. В дальнейшем (с возрастом) у лиц с подобными нарушениями высока вероятность развития деменции или состояний, близких к синдрому Альцгеймера.
На 79 конгрессе Европейского общества по изучению атеросклероза в Швеции (2011) испанские исследователи [М. Solancs et al.] сообщили о двух мутациях, которые они обнаружили у 9 обследованных с III типом ГЛП (они обозначили их как R136S и AL149).
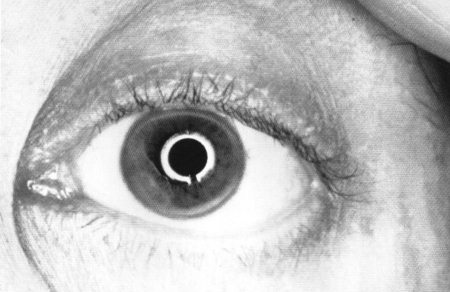
Рис. 3. Липоидные дуги роговицы

Рис. 4. Ксантелазмы
Из моногенных мутаций чаще всего приходится сталкиваться с семейными ГХС, связанными с дефицитом рецепторов ЛПНП или нарушениями структуры самих ЛПНП-частиц.
Приводим характерные признаки клинических проявлений ГХС. На рисунках 3 и 4 видны липоидные дуги роговицы (верхний рисунок) и ксантелазмы (нижний рисунок).
На рисунках 5 и 6 показаны сухожильные ксантомы пястно-фаланговых суставов и ахилловых сухожилий.
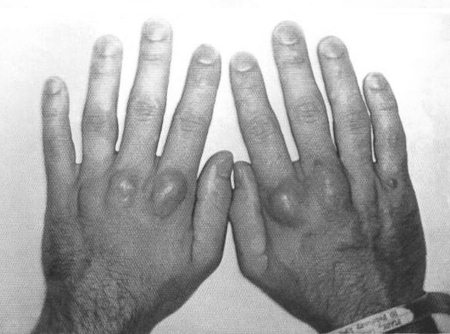
Рис. 5. Сухожильные ксантомы пястно-фаланговых суставов

Рис. 6. Сухожильные ксантомы ахилловых сухожилий
Для наших широт наиболее типична семейная ГХС по причине дефицита или дефекта рецепторов ЛПНП в печеночных и других соматических клетках или их низкой функциональной активности (в связи с дефектом структуры). Следствие этого – накопление ЛПНП-частиц – основных носителей ХС – в циркулирующей крови.
При относительно частых гетерозиготных формах семейной ГХС (1 случай на 500 человек населения) уровень ХС крови у этих лиц обычно колеблется в пределах 8-10 ммоль/л (350–500 мг/дл). В редких случаях гомозиготной семейной ГХС (1 случай на 1 млн жителей) уровень ХС крови достигает 15–20 ммоль/л (650-1000 мг/дл).
Нуклеотидная цепь гена рецептора ЛПНП состоит примерно из 45 тысяч нуклеотидов. Надо полагать, что при ее построении вероятность «ошибки» или мутации довольно велика, причем эти «сбои» могут возникать в разных участках нуклеотидной цепи, способны «закрепляться» и наследоваться. Описано около 1500 вариантов мутаций, ведущих к семейной ГХС и вызывающих нарушение работы рецепторов ЛПНП.
Один из вариантов этой мутации довольно широко распространен среди восточно-европейских евреев – «ашкенази», предки которых были выходцами из Литвы, из-за чего мутация получила название «литовской». Интересно, что среди евреев испанского происхождения – давних жителей Средиземноморья (евреев-«сефардов») – «литовская» мутация не встречается.
Среди франко-канадцев и жителей ЮАР с голландскими корнями частота этого заболевания очень высока (1 случай на 100–300 человек), что получило название «эффекта основателя». Это объясняют тем, что в относительно ограниченной популяции заключаются в основном только «внутренние» браки, т. е. эта этническая группа почти не смешивается с более широкими слоями населения, что увеличивает шанс появления генетически обусловленной патологии.
Мутации семейной ГХС, связанные с дефектом гена рецепторов ЛПНП, возникают в одном из генетических локусов 19-й хромосомы.
Другой вариант семейной ГХС характеризуется нарушением структуры апопротеина В-100 – ключевого белка ЛПНП. В этом случае нормальные рецепторы ЛПНП плохо распознают ЛПНП и плохо связываются с ними, поскольку апо В-100 является лигандом (связующим звеном) рецепторов ЛПНП. Ген, контролирующий структуру апо В-100, локализуется во 2-й хромосоме. Мутация этого гена влечет за собой дефект апо В-100, что приведет к плохой эвакуации ЛПНП из циркулирующей крови. Это другой вариант семейной ГХС. Данный тип мутации чаще всего встречается среди народов, населяющих страны Западной Европы.
Оба описанных варианта семейной ГХС имеют характер доминантных мутаций.
Описан третий (редкий) вид семейной ГХС, который носит аутосомно-рецессивный характер (ARH). Мутация была обнаружена в одной ливанской семье и получила название «ливанской». Мутация локализуется в одном из генов 1-й хромосомы. Этот ген «отвечает» за синтез транспортного белка, переносящего комплекс – рецептор ЛПНП + ЛПНП – с поверхности печеночных клеток внутрь, к органоидам, которые должны утилизировать ЛПНП. Если указанный транспортный белок не способен переносить комплексы рецептор ЛПНП+ ЛПНП внутрь клетки, они скапливаются на мембране и блокируют дальнейшее связывание ЛПНП. Следствием этого будет задержка ЛПНП в периферической крови, т. е. ГХС.
Недавно открыт еще один – 4-й – вид моногенной аутосомно-доминантной мутации, тоже приводящей к семейной ГХС. Это результат мутации одного из генов 1-й хромосомы, контролирующего синтез фермента «пропротеин-конвертазы субтилизин-кексинового типа 9», обозначаемого как PCSK-9. В зависимости от особенностей своей структуры этот фермент может либо тормозить, либо стимулировать активность рецепторов ЛПНП. В том случае, когда продуцируется PCSK-9 с повышенным сродством к рецепторам ЛПНП, деятельность этих рецепторов будет угнетаться, что приведет к ГХС.
Интересно, что мутация этого фермента может оказаться и прямо противоположной той, что описана выше. В результате этого сродство PCSK-9 к рецепторам ЛПНП понизится, они (рецепторы ЛПНП) будут весьма активно работать, что вызовет снижение уровня ЛПНП и ХС крови. В этом случае можно говорить о мутации с положительным знаком, которая обеспечит устойчивость к развитию атеросклероза.
Известна также моногенная мутация, связанная не с ГЛП, а с дислипидемией (ДЛП) без ГХС, приводящая к распространенному атеросклерозу из-за очень низкого содержания в плазме крови антиатерогенной фракции – ЛПВП. Это редкое эндемичное заболевание – Танжерская болезнь, которая распространена на одном из островов в Западной Атлантике. Заболевание развивается вследствие гомозиготной мутации гена, локализованного в 9-й хромосоме и кодирующего синтез транспортного белка АВСА-1. Этот белок выполняет функцию специализированного внутриклеточного переносчика свободного ХС к ключевому апопротеину ЛПВП – апо А1. Если в организме возникает дефицит АВСА, блокируется образование зрелых, функционально активных форм ЛПВП. Помимо нехватки транспортного белка (АВСА-1), при Танжерской болезни значительно снижается также содержание апобелков А-1 и A-II – ключевых белков ЛПВП, без которых их синтез невозможен.
Дефицит ЛПВП-3 и ЛПВП-2 резко снижает обратный транспорт ХС с поверхности периферических клеток в печень. ХС задерживается в тканях, в том числе накапливается в сосудистой стенке, что ведет к образованию атеросклеротических бляшек, хотя ГХС при этом не развивается.
В литературе описаны и другие гомозиготные мутации, связанные с дефектами других белковых транспортеров, переносящих стерины, – это ABCG-5, ABCG-8. При нарушенной структуре этих белков выход стеринов из клеток резко затрудняется [К. Berge et al., 2000].
Как правило, такие дефекты сопровождаются повышенным всасыванием растительных стеринов [J. Horton et al., 2002], что приводит к сито-стеролемии, хотя ГХС отсутствует. Поскольку необходимых белковых переносчиков стеринов мало, в организме нарушается способность выделять стерины, которые постепенно накапливаются в печени. Это влечет за собой подавление синтеза рецепторов ЛПНП, что в дальнейшем тоже может вызвать ГХС.
Дислипидемические состояния, характеризующиеся избирательным снижением уровня ЛПВП (без ГХС и без ГТГ) встречаются довольно часто во всех регионах мира, хотя они не связаны с Танжерской болезнью. При гетерозиготном носительстве дефектного гена АВСА-1 Танжерская болезнь не развивается, но содержание ЛПВП-фракции бывает отчетливо сниженным, что делает таких людей подверженными атеросклерозу [М.Ю. Мандельштам, В. Б. Васильев, 2008]. Даже один патологический аллель гена, управляющего синтезом белка АВСА-1, может оказать отрицательное влияние на этот транспортный белок и сделать его недостаточно функциональным.
В некоторых случаях дефицит ЛПВП развивается как результат аномалии в строении генов, контролирующих синтез лецитин-холестерин-ацилтрансферазы (ЛХАТ), участие которой необходимо для эстерификации свободного ХС. Этот дефект возникает как результат полигенной патологии при комбинации нескольких функционально значимых нуклеотидных полиморфизмов (в нескольких генетических локусах).
Еще один моногенный (генетически обусловленный) рецессивный дефект – причина редкой формы дислипидемии – абеталипопротеинемии. При этом типе ДЛП нарушается синтез апопротеина В, резко тормозится образование ЛПНП-частиц и снижается уровень ХС и ТГ крови. Хотя в этом случае исчезает почва для развития атеросклероза, развиваются серьезные нарушения со стороны центральной и периферической нервной системы, поскольку при крайнем дефиците ЛПНП в первую очередь страдает трофика нервных клеток и их отростков.
Из других мало известных моногенных дефектов, связанных с развитием ДЛП, можно указать на мутацию гена, управляющего продукцией скевенджер-рецепторов. Функция этих клеточных образований печени заключается в том, чтобы захватывать возвращающиеся в печень ЛПВП, нагруженные ХС, который они принесли с периферии. Если функция скевенджер-рецепторов нарушена, ЛПВП плохо проникают в печень, их концентрация в периферической крови повышается, но функция парализуется, т. к. они нагружены ХС до предела и больше не могут выполнять свою антиатерогенную миссию [D. Osgood et al., 2003]. Уровень общего ХС крови при этом повышается. Таким образом, иногда приходится сталкиваться с больными, у которых ЛПВП много, но функционально они неактивны. Иначе говоря, встречаются индивиды с высоким содержанием ЛПВП-частиц, а их защищенность от атеросклероза находится на очень низком уровне.
Похожая ситуация возникает при генетически обусловленной недостаточности триглицеридлипазы, которая плохо расщепляет ТГ, не освобождает от них ЛПВП, что резко тормозит антиатерогенную функцию этих частиц [R. Hegele et al., 1991].
Плохая работоспособность ЛПВП может быть связана не только с их дефицитом, но с генетически обусловленной патологией (недостатком) переносящих липиды транспортных белков. Так, при недостатке такого транспортного белка, как СЕТР (переносит ХС-эстеры и ТГ), затрудняется мобилизация эстерифицированного ХС из ЛПВП, эти частицы укрупняются, перегружаются ХС и вместо акцепторов ХС становятся его донорами [ILLUMINATE, 2006]. При этом концентрация ХС ЛПВП в периферической крови доходит до 70 мг/дл (1,8 ммоль/л) и выше. На этом примере еще раз можно убедиться в том, что не всегда высокое содержание антиатерогенной фракции липопротеидов (ЛПВП) в циркулирующей крови гарантирует защиту от атеросклероза. Одного такого больного мы наблюдали.
Приводим наше наблюдение № 1.
Больной С., 65 лет (1995 г.). В марте 1995 г. больной перенес острый инфаркт миокарда на переднебоковой стенке левого желудочка. При осмотре в домашних условиях через 3 дня после сердечного приступа (больной отказался от госпитализации) состояние больного удовлетворительное, пульс – 60 уд/мин, АД= 140/80 мм рт. ст. В анамнезе – много лет АГ, нерегулярно принимал гипотензивные препараты. В 1980 г. (в возрасте 50 лет) перенес первый инфаркт миокарда. В мае 1995 г. при исследовании липидного состава крови общий ХС = 308 мг/дл (7,9 ммоль/л), ТТ=185 мг/дл (2,1 ммоль/л), ХС ЛПВП = 91 мг/дл (2,3 ммоль/л), ХС ЛПНП= 180 мг/дл (4,6 ммоль/л), коэффициент атерогенности – 2,4 ед.
РЕЗЮМЕ. Несмотря на очень высокий уровень ХС ЛПВП (91 мг/дл) и нормальный коэффициент атерогенности, больной дважды перенес инфаркт миокарда. Есть все основания предполагать, что ЛПВП-частицы у данного больного не выполняют предназначенную им роль. Этих частиц много, но функционально они, по-видимому, неполноценны.
Встречаются и другие формы генетически детерминированных дислипидемий, например, комбинированная гиперлипидемия, при которой повышен уровень и ХС, и ТГ. Этот вид ДЛП (ГЛП) возникает в результате мутации гена, контролирующего синтез транскрипционной ДНК, которая описана под названием Upstream Stimulatory Factor (USF-1). Это также моногенная мутация, при которой главным образом нарушается элиминация ЛПОНП, а значит и ТГ, из циркулирующей крови. Впервые данная мутация была идентифицирована в Финляндии и в своем гетерозиготном варианте, по-видимому, обусловливает ГТГ у лиц с метаболическим синдромом. Авторы, описавшие эту мутацию [P. Pajukanta et al., 2004], рассматривают USF-1 как регулятор работы других генов, который посредством РНК кодирует синтез ряда белков – участников обмена и транспортировки апопротеинов А-2, С-3, Е, АВСА-1 и др.
Особого внимания требует оценка генетической детерминации апопротеина Е. По данным исследователей из Южной Кореи [Н. Oh et al., 2008], установлено несколько изоформ апоЕ (апо Е-2, апо Е-3, апо Е-4), которые могут встречаться в разных комбинациях, что зависит от наследственно обусловленного генотипа. Было установлено, что у большинства практически здоровых лиц, не имеющих ДЛП, обычно встречается генотип Е-З/Е-З (гомозиготный) или аллели Е-З/Е-2 либо Е-З/Е-4 (гетерозиготные). В некоторых случаях (из-за наличия определенных нуклеотидных полиморфизмов в каких-то генах) образуются генотипы Е-2/Е-2 или Е4/Е4, при которых продуцируются неполноценные апопротеины Е [D. Betteridge & J. Morrell, 2003]. В части таких случаев возникает предрасположенность к развитию тяжелой комбинированной гиперлипидемии, близкой к III типу по классификации Фредриксона. В дальнейшем (с возрастом) у лиц с подобными нарушениями высока вероятность развития деменции или состояний, близких к синдрому Альцгеймера.
На 79 конгрессе Европейского общества по изучению атеросклероза в Швеции (2011) испанские исследователи [М. Solancs et al.] сообщили о двух мутациях, которые они обнаружили у 9 обследованных с III типом ГЛП (они обозначили их как R136S и AL149).
Рис. 3. Липоидные дуги роговицы
Рис. 4. Ксантелазмы
Из моногенных мутаций чаще всего приходится сталкиваться с семейными ГХС, связанными с дефицитом рецепторов ЛПНП или нарушениями структуры самих ЛПНП-частиц.
Приводим характерные признаки клинических проявлений ГХС. На рисунках 3 и 4 видны липоидные дуги роговицы (верхний рисунок) и ксантелазмы (нижний рисунок).
На рисунках 5 и 6 показаны сухожильные ксантомы пястно-фаланговых суставов и ахилловых сухожилий.
Рис. 5. Сухожильные ксантомы пястно-фаланговых суставов
Рис. 6. Сухожильные ксантомы ахилловых сухожилий
1.4.2. Полигенные ДЛП
Популяционные обследования, нацеленные на выявление дислипидемических состояний, в основном выявляют первичные ДЛП, имеющие характер полигенных, т. е. вызванных генетически обусловленными нуклеотидными полиморфизмами, создающими наследственное предрасположение к этим состояниям.
Обследование взрослого населения городов европейской части России выявило 15–20 % индивидов с такими нарушениями, которые реализуются в условиях дополнительных неблагоприятных факторов внешней среды, например, это может быть рацион, содержащий много животных жиров, сахаристых продуктов; малоподвижный образ жизни; курение; стрессы и пр.
Надо признать, что контролировать липидный состав крови с помощью диеты и лекарственных препаратов значительно легче при полигенных ДЛП, тогда как моногенные ДЛП труднее поддаются терапевтическому воздействию.
Что касается проявлений полигенных ДЛП, то чаще всего это умеренная ГХС или гипертриглицеридемии (ГТГ в пределах 2,0–2,5 ммоль/л).
Если патологическое значение ГХС в настоящее время ни у кого не вызывает сомнений, то в отношении вредного воздействия умеренного повышения уровня ТГ крови единой точки зрения нет. В современной литературе все больше работ, в которых придается патологическое значение постпрандиальной ГТГ, т. е. повышению концентрации ТГ в плазме крови через 1–2 часа после еды. Надо иметь в виду, что главные носители ТГ – ЛПОНП, которые являются основным субстратом для продукции ЛПНП – главного виновника процесса инфильтрации липидов в сосудистую стенку после своей модификации.
Установлены реципрокные взаимоотношения между ТГ и ЛПОНП, с одной стороны, и ЛПВП, с другой. Чем выше уровень ЛПОНП, тем ниже содержание ЛПВП. Недаром в последних руководствах по липидологии предлагается говорить не столько о ХС ЛПНП, как показателе риска атеросклероза, сколько о ХС, не связанном с ЛПВП. Этот ХС рассчитывается как разность между общим ХС и суммой ХС ЛПОНП, ХС ЛП промежуточной плотности (ХС ЛППП) и ХС ЛПНП. Еще проще этот показатель атерогенного ХС рассчитывать как разницу между общим ХС и ХС ЛПВП [ESC/EAS Guidelines, 2011]. Именно этот ХС является опорным критерием для оценки патологического значения ГХС.
Обследование взрослого населения городов европейской части России выявило 15–20 % индивидов с такими нарушениями, которые реализуются в условиях дополнительных неблагоприятных факторов внешней среды, например, это может быть рацион, содержащий много животных жиров, сахаристых продуктов; малоподвижный образ жизни; курение; стрессы и пр.
Надо признать, что контролировать липидный состав крови с помощью диеты и лекарственных препаратов значительно легче при полигенных ДЛП, тогда как моногенные ДЛП труднее поддаются терапевтическому воздействию.
Что касается проявлений полигенных ДЛП, то чаще всего это умеренная ГХС или гипертриглицеридемии (ГТГ в пределах 2,0–2,5 ммоль/л).
Если патологическое значение ГХС в настоящее время ни у кого не вызывает сомнений, то в отношении вредного воздействия умеренного повышения уровня ТГ крови единой точки зрения нет. В современной литературе все больше работ, в которых придается патологическое значение постпрандиальной ГТГ, т. е. повышению концентрации ТГ в плазме крови через 1–2 часа после еды. Надо иметь в виду, что главные носители ТГ – ЛПОНП, которые являются основным субстратом для продукции ЛПНП – главного виновника процесса инфильтрации липидов в сосудистую стенку после своей модификации.
Установлены реципрокные взаимоотношения между ТГ и ЛПОНП, с одной стороны, и ЛПВП, с другой. Чем выше уровень ЛПОНП, тем ниже содержание ЛПВП. Недаром в последних руководствах по липидологии предлагается говорить не столько о ХС ЛПНП, как показателе риска атеросклероза, сколько о ХС, не связанном с ЛПВП. Этот ХС рассчитывается как разность между общим ХС и суммой ХС ЛПОНП, ХС ЛП промежуточной плотности (ХС ЛППП) и ХС ЛПНП. Еще проще этот показатель атерогенного ХС рассчитывать как разницу между общим ХС и ХС ЛПВП [ESC/EAS Guidelines, 2011]. Именно этот ХС является опорным критерием для оценки патологического значения ГХС.
1.4.3. Клинические проявления дислипидемий
Наиболее тяжелые гомозиготные моногенные формы гиперхолестеринемии чаще всего начинают проявляться с 5–7 лет, когда у ребенка возникают желтоватые, возвышающиеся над кожей пятна – ксантомы – на разгибательных поверхностях рук, в области локтей, а иногда и в других участках кожи, например в ягодичной области. В более старшем возрасте формируются сухожильные ксантомы в области пястно-фаланговых суставов и ахилловых сухожилий (рис. 5 и 6).
Конец бесплатного ознакомительного фрагмента