Страница:
Ученые-иммунологи предполагают, что вследствие генной мутации нарушается синтез цепей иммуноглобулинов. Обычно мутации подвергается ген-регулятор, локализующийся в Х-хромосоме, в результате этого нарушается синтез иммуноглобулинов.
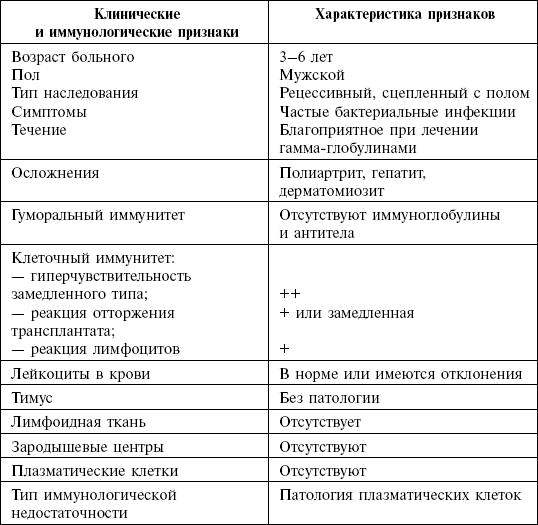
У больных снижены показатели уровня JgG (менее 15 г/л), JgM, JgA, В-лимфоциты обычно отсутствуют в связи с отсутствием тирозинкеназы, необходимой для их созревания (табл. 1).
Лечение проводится иммуноглобулином в течение всей жизни больного.
Таблица 1. Клиническая и иммунологическая характеристика первичной агаммаглобулинемии Брутона
Селективный дефицит иммуноглобулина А
Синдром Ди Джорджи
Хронический слизисто-кожный кандидоз
Алимфоцитоз (лимфоцитарная дисгенезия, лимфоцитарная аплазия, французский тип иммунопареза, синдром Незелофа)
Швейцарский тип агаммаглобулинемии
Иммунный дефицит с тромбоцитопенией и экземой (синдром Вискотта – Олдрича)
Синдром Луи – Бар (атаксия-телеангиэктазия)
Синдром Чедиака – Хигаси – Штайнбринка
Синдром Гуда (тимома)
Хроническая гранулематозная болезнь
Наследственный ангионевротический отек
Глава 4
Диагностика
Лечение
Глава 5
Лечение вторичных иммунодефицитов
Глава 6
Правила подготовки пациентов к обследованию, взятие и условия хранения материала для проведения исследования
Некоторые методы исследования иммунного статуса
У больных снижены показатели уровня JgG (менее 15 г/л), JgM, JgA, В-лимфоциты обычно отсутствуют в связи с отсутствием тирозинкеназы, необходимой для их созревания (табл. 1).
Лечение проводится иммуноглобулином в течение всей жизни больного.
Таблица 1. Клиническая и иммунологическая характеристика первичной агаммаглобулинемии Брутона
Селективный дефицит иммуноглобулина А
Изолированный дефицит иммуноглобулина А диагностируется одновременно с повышенным или нормальным уровнем других иммуноглобулинов. Это может обнаруживаться у здоровых лиц при различных обследованиях. В большинстве случаев отсутствие JgA сочетается с хромосомными аномалиями 18-й пары хромосом.
Основные клинические симптомы разнообразны. Наиболее часто встречаются пульмональные инфекции, атонии, жидкий стул и аутоиммунные заболевания. Поражения пищеварительной и дыхательной систем объясняются отсутствием секреторного компонента JgA. Больные имеют склонность к образованию иммунных комплексов.
Селективный дефицит JgA выявлен при красной волчанке, сахарном диабете, болезни Аддисона, хроническом гепатите, тромбоцитопенической пурпуре, гемолитической анемии.
Лечение проводится иммуноглобулином с высоким содержанием JgA.
Однако при этом отмечаются побочные реакции, возникающие в результате выработки антител JgG против экзогенного JgA. В этих случаях применяется препарат Гаммакард (5 %-й или 10 %-й раствор).
Основные клинические симптомы разнообразны. Наиболее часто встречаются пульмональные инфекции, атонии, жидкий стул и аутоиммунные заболевания. Поражения пищеварительной и дыхательной систем объясняются отсутствием секреторного компонента JgA. Больные имеют склонность к образованию иммунных комплексов.
Селективный дефицит JgA выявлен при красной волчанке, сахарном диабете, болезни Аддисона, хроническом гепатите, тромбоцитопенической пурпуре, гемолитической анемии.
Лечение проводится иммуноглобулином с высоким содержанием JgA.
Однако при этом отмечаются побочные реакции, возникающие в результате выработки антител JgG против экзогенного JgA. В этих случаях применяется препарат Гаммакард (5 %-й или 10 %-й раствор).
Синдром Ди Джорджи
Этот синдром возникает при врожденной аплазии тимуса на 6–10-й неделе гестации в результате порока развития 3-го и 4-го глоточных карманов, из которого развиваются паращитовидная и зобная железы. Они подвергнуты гипоплазии или находятся в полной аплазии. Кроме этого могут поражаться и соседние органы, что проявляется в виде дистрофии лица, гипоплазии нижней челюсти, короткой верхней губы, антимонголоидно расположенными глазными щелями, низким расположением ушных раковин.
Этому заболеванию в большей степени подвержены девочки. В период новорожденности заболевание может проявляться тетанией, гипокальциемией, кандидамикозом.
Часто синдром Ди Джорджи сочетается с врожденными пороками крупных сосудов и сердца (общий артериальный проток, двойная дуга аорты, декстракардия и др.).
Клиническое проявление синдрома фиксируется сразу после рождения в виде дистрофии лица, тогда же устанавливаются врожденные пороки сердца.
В неонатальном периоде наиболее частым симптомом являются гипокальциемические судороги. Иммунодефицитный синдром развивается во втором полугодии жизни грудного ребенка и проявляется рецидивирующими инфекциями, вызванными вирусами, бактериями, условно-патогенными микробами, грибками.
Симптомы иммунодефицита различаются в зависимости от степени поражения зобной железы. Отмечаются дефекты дифференциации стволовых клеток в Т-клетки. В большинстве случаев аплазия тимуса неполная и функция Т-лимфоцитов может восстанавливаться.
В подобных случаях эффективна трансплантация тимуса плода.
Существует несколько способов трансплантации тимуса в мышцу передней брюшной стенки, внутривенное или внутрибрюшное введение клеток тимуса, внутрибрюшное введение небольших фрагментов тимуса.
Этому заболеванию в большей степени подвержены девочки. В период новорожденности заболевание может проявляться тетанией, гипокальциемией, кандидамикозом.
Часто синдром Ди Джорджи сочетается с врожденными пороками крупных сосудов и сердца (общий артериальный проток, двойная дуга аорты, декстракардия и др.).
Клиническое проявление синдрома фиксируется сразу после рождения в виде дистрофии лица, тогда же устанавливаются врожденные пороки сердца.
В неонатальном периоде наиболее частым симптомом являются гипокальциемические судороги. Иммунодефицитный синдром развивается во втором полугодии жизни грудного ребенка и проявляется рецидивирующими инфекциями, вызванными вирусами, бактериями, условно-патогенными микробами, грибками.
Симптомы иммунодефицита различаются в зависимости от степени поражения зобной железы. Отмечаются дефекты дифференциации стволовых клеток в Т-клетки. В большинстве случаев аплазия тимуса неполная и функция Т-лимфоцитов может восстанавливаться.
В подобных случаях эффективна трансплантация тимуса плода.
Существует несколько способов трансплантации тимуса в мышцу передней брюшной стенки, внутривенное или внутрибрюшное введение клеток тимуса, внутрибрюшное введение небольших фрагментов тимуса.
Хронический слизисто-кожный кандидоз
Синдром наследуется по аутосомно-рецессивному типу. В этих случаях Candida albicans вызывает поражение слизистой рта и влагалища, кожи, волосистой части головы, ногтей. У таких больных встречаются аутоиммунные нарушения с поражением надпочечников, щитовидной железы.
При исследовании определяется нормальный уровень иммуноглобулинов со снижением JgA, причем число и функции Т-лимфоцитов не нарушены, за исключением реакции Т-лимфоцитов на грибковые антигены.
В качестве лечения применяют противогрибковые препараты, проводят трансплантации клеток тимуса, назначают прием его экстрактов.
При исследовании определяется нормальный уровень иммуноглобулинов со снижением JgA, причем число и функции Т-лимфоцитов не нарушены, за исключением реакции Т-лимфоцитов на грибковые антигены.
В качестве лечения применяют противогрибковые препараты, проводят трансплантации клеток тимуса, назначают прием его экстрактов.
Алимфоцитоз (лимфоцитарная дисгенезия, лимфоцитарная аплазия, французский тип иммунопареза, синдром Незелофа)
Этот синдром относится к наследственной недостаточности иммунитета и характеризуется отсутствием реакций иммунологической защиты.
При этом заболевании отмечается количественная и качественная недостаточность лимфоцитов при нормальном содержании лимфоцитов в плазме крови, кроме того, поражаются Т-лимфоциты, однако отмечается нормальное количество В-лимфоцитов.
Заболевание наследуется по аутосомно-рецессивному типу, проявляется в раннем детском возрасте и характеризуется злокачественным течением.
У ребенка отмечается задержка роста, развитие септического процесса с появлением гнойных очагов в коже, легких и других органах, может развиться грибковый сепсис. В периферической крови происходит довольно резкое снижение содержания лимфоцитов, слабо выражена реакция бластной трансформации лимфоцитов.
Иммуноглобулины всех классов в периферической крови находятся на нормальном уровне. Это заболевание в большинстве случаев заканчивается летальным исходом в связи с развитием септического состояния.
Кроме комплексного лечения, в этих случаях рекомендуется пересадка костного мозга.
При этом заболевании отмечается количественная и качественная недостаточность лимфоцитов при нормальном содержании лимфоцитов в плазме крови, кроме того, поражаются Т-лимфоциты, однако отмечается нормальное количество В-лимфоцитов.
Заболевание наследуется по аутосомно-рецессивному типу, проявляется в раннем детском возрасте и характеризуется злокачественным течением.
У ребенка отмечается задержка роста, развитие септического процесса с появлением гнойных очагов в коже, легких и других органах, может развиться грибковый сепсис. В периферической крови происходит довольно резкое снижение содержания лимфоцитов, слабо выражена реакция бластной трансформации лимфоцитов.
Иммуноглобулины всех классов в периферической крови находятся на нормальном уровне. Это заболевание в большинстве случаев заканчивается летальным исходом в связи с развитием септического состояния.
Кроме комплексного лечения, в этих случаях рекомендуется пересадка костного мозга.
Швейцарский тип агаммаглобулинемии
Заболевание относится к смешанному типу иммунодефицитного состояния, при котором нарушены как клеточные, так и гуморальные типы иммунологической защиты, является редкой формой нарушения иммунитета.
Заболевание проявляется на 2–3-м месяце жизни и отмечается злокачественным течением с поражением бронхолегочной системы и желудочно-кишечного тракта. Наследование может быть рецессивным, сцепленным с Х-хромосомой, или аутосомно-рецессивным.
Иммунологические нарушения выражаются в снижении продукции циркулирующих антител, снижении или дефиците всех типов иммуноглобулинов, неспособности проявлять реакции замедленной гиперчувствительности и отторгать кожный трансплантат.
При швейцарском типе агаммаглобулинемии отсутствуют миндалины, аденоиды и пейеровы бляшки, может отмечаться дисплазия, гипоплазия или рудиментарный тимус. При швейцарском типе агаммаглобулинемии возможен летальный исход в первые месяцы жизни ребенка.
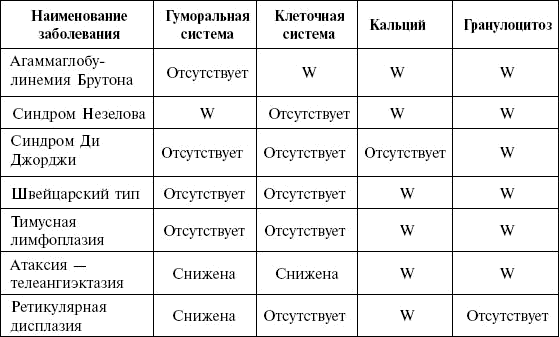
В табл. 2 представлены отличительные признаки различных видов агаммаглобулинемии.
Таблица 2. Сравнительный анализ признаков различных видов агаммаглобулинемии
Заболевание проявляется на 2–3-м месяце жизни и отмечается злокачественным течением с поражением бронхолегочной системы и желудочно-кишечного тракта. Наследование может быть рецессивным, сцепленным с Х-хромосомой, или аутосомно-рецессивным.
Иммунологические нарушения выражаются в снижении продукции циркулирующих антител, снижении или дефиците всех типов иммуноглобулинов, неспособности проявлять реакции замедленной гиперчувствительности и отторгать кожный трансплантат.
При швейцарском типе агаммаглобулинемии отсутствуют миндалины, аденоиды и пейеровы бляшки, может отмечаться дисплазия, гипоплазия или рудиментарный тимус. При швейцарском типе агаммаглобулинемии возможен летальный исход в первые месяцы жизни ребенка.
В табл. 2 представлены отличительные признаки различных видов агаммаглобулинемии.
Таблица 2. Сравнительный анализ признаков различных видов агаммаглобулинемии
Иммунный дефицит с тромбоцитопенией и экземой (синдром Вискотта – Олдрича)
Для данного синдрома характерно наследование по рецессивному, то есть сцепленному с Х-хромосомой, типу.
Заболевание диагностируется у мальчиков в раннем возрасте. Этот синдром характеризуется наличием трех основных симптомов:
– тромбоцитопенией, приводящей к носовым кровотечениям;
– экземой;
– склонностью к гнойным инфекциям (дыхательные пути, отиты).
Заболевание проявляется в период новорожденности в виде кровоизлияний на коже и кровавой диареи. Спустя некоторое время появляются носовые кровотечения. В течение первых 3 месяцев жизни появляются экзема, напоминающая атопический дерматит и уртикарная сыпь с эозинофилией. В первом полугодии частыми являются тяжелые инфекции (сепсис, менингиты и др.), с возрастом явления иммунного дефицита уменьшаются.
При лабораторных исследованиях у такого рода детей определяется низкий уровень иммуноглобулина М при нормальном содержании JgG и повышенном JgA, выявляется лимфоцитопения, низкая активность Т-лимфоцитов на полисахаридные антигены.
При данном заболевании требуется контроль свертывающей системы крови. Обычно назначают переливание эритроцитарной и тромбоцитарных концентратов.
На экзему воздействуют применением гормональных препаратов. Присоединившиеся инфекционные заболевания лечат специфическими антибиотиками.
Пересадка костного мозга способствует достижению положительных результатов в лечении и существенно увеличивает продолжительность жизни больных.
Заболевание диагностируется у мальчиков в раннем возрасте. Этот синдром характеризуется наличием трех основных симптомов:
– тромбоцитопенией, приводящей к носовым кровотечениям;
– экземой;
– склонностью к гнойным инфекциям (дыхательные пути, отиты).
Заболевание проявляется в период новорожденности в виде кровоизлияний на коже и кровавой диареи. Спустя некоторое время появляются носовые кровотечения. В течение первых 3 месяцев жизни появляются экзема, напоминающая атопический дерматит и уртикарная сыпь с эозинофилией. В первом полугодии частыми являются тяжелые инфекции (сепсис, менингиты и др.), с возрастом явления иммунного дефицита уменьшаются.
При лабораторных исследованиях у такого рода детей определяется низкий уровень иммуноглобулина М при нормальном содержании JgG и повышенном JgA, выявляется лимфоцитопения, низкая активность Т-лимфоцитов на полисахаридные антигены.
При данном заболевании требуется контроль свертывающей системы крови. Обычно назначают переливание эритроцитарной и тромбоцитарных концентратов.
На экзему воздействуют применением гормональных препаратов. Присоединившиеся инфекционные заболевания лечат специфическими антибиотиками.
Пересадка костного мозга способствует достижению положительных результатов в лечении и существенно увеличивает продолжительность жизни больных.
Синдром Луи – Бар (атаксия-телеангиэктазия)
Этот синдром является комплексным заболеванием иммунной, нервной и эндокринной систем. Наследуется по аутосомнорецессивному типу и является типичным симптомом хромосомных разрывов из-за чувствительности к повреждающим агентам ДНК.
Основные клинические симптомы развиваются после первого года жизни. К ним относятся:
– мозжечковая атаксия;
– телеангиэктазия конъюнктивы, кожи и слизистых оболочек ротовой полости;
– рецидивирующие инфекции легких, дыхательных путей.
Мозжечковая атаксия является наиболее характерным признаком заболевания, наступает обычно в школьном возрасте. Телеангиэктазии проявляют себя в возрасте 3–6 лет: поражаются конъюнктивы, определяются расширение и извитие малых вен. Подобное расширение вен происходит и на ушных раковинах, и щеках. Помимо телеангиэктазий возможно образование кожных пятен цвета «кофе с молоком».
Кожа выглядит преждевременно постаревшей, часто встречается поседение волос в пубертатном периоде, обнаруживается склонность к инфекциям в виде поражения дыхательных путей, в виде синопульмональных воспалений. Кроме основных симптомов появляются эндокринологические расстройства: гипоплазии, аплазии яичников, снижение толерантности к глюкозе, низкорослость.
Часто повышается инсулин в плазме, возникает диабет, резистентный к инсулину, так как вырабатываются антитела против рецепторов инсулина. Развивается гипоплазия и аплазия тимуса, наиболее частой иммунологической аномалией являются отсутствие или низкое содержание JgA и JgЕ, низкое число Т-лимфоцитов.
Концентрация JgM нормальная или повышена.
У большинства больных обнаруживаются признаки нарушения клеточного иммунитета.
Общее число лимфоцитов уменьшено незначительно, при значительном понижении Т-клеток.
В качестве лечения проводится антибактериальная терапия. Внутривенно вводится иммуноглобулин.
Прогноз неблагоприятный, обычно иммунологическая коррекция оказывается неэффективной.
Основные клинические симптомы развиваются после первого года жизни. К ним относятся:
– мозжечковая атаксия;
– телеангиэктазия конъюнктивы, кожи и слизистых оболочек ротовой полости;
– рецидивирующие инфекции легких, дыхательных путей.
Мозжечковая атаксия является наиболее характерным признаком заболевания, наступает обычно в школьном возрасте. Телеангиэктазии проявляют себя в возрасте 3–6 лет: поражаются конъюнктивы, определяются расширение и извитие малых вен. Подобное расширение вен происходит и на ушных раковинах, и щеках. Помимо телеангиэктазий возможно образование кожных пятен цвета «кофе с молоком».
Кожа выглядит преждевременно постаревшей, часто встречается поседение волос в пубертатном периоде, обнаруживается склонность к инфекциям в виде поражения дыхательных путей, в виде синопульмональных воспалений. Кроме основных симптомов появляются эндокринологические расстройства: гипоплазии, аплазии яичников, снижение толерантности к глюкозе, низкорослость.
Часто повышается инсулин в плазме, возникает диабет, резистентный к инсулину, так как вырабатываются антитела против рецепторов инсулина. Развивается гипоплазия и аплазия тимуса, наиболее частой иммунологической аномалией являются отсутствие или низкое содержание JgA и JgЕ, низкое число Т-лимфоцитов.
Концентрация JgM нормальная или повышена.
У большинства больных обнаруживаются признаки нарушения клеточного иммунитета.
Общее число лимфоцитов уменьшено незначительно, при значительном понижении Т-клеток.
В качестве лечения проводится антибактериальная терапия. Внутривенно вводится иммуноглобулин.
Прогноз неблагоприятный, обычно иммунологическая коррекция оказывается неэффективной.
Синдром Чедиака – Хигаси – Штайнбринка
Заболевание наследуется по аутосомно-рецессивному типу.
Характерными клиническими симптомами являются:
– альбинизм;
– обесцвечивание радужки глаза;
– гепатоспленомегалия;
– увеличение периферических лимфоузлов;
– рецидивирующие вирусные и кишечные инфекции;
– периферические нейропатии.
Наиболее характерным является повышенная чувствительность к гнойным инфекциям, гипопигментации кожи, волос, радужной оболочки глаз.
Поражения желудочно-кишечного тракта начинаются уже в период новорожденности, отмечается повышенная чувствительность кожи больных к солнечному свету.
Дефект пигментации связан с нарушением распределения клеток, несущих меланин коже и волосам больных, а также с недостаточной деструкцией меланосом, которые обычно располагаются аномально. В связи с этим цвет кожи варьируется от кремового до серого, цвет волос – блондин или брюнет с серым оттенком.
Со стороны крови определяется нейтропения, тромбоцитопения, анемия.
В лейкоцитах обнаруживаются гигантские включения, дающие положительную реакцию на пероксидазу; в цитоплазме нейтрофилов, моноцитов, лимфоцитов – гиганты-гранулы, содержащие миелопероксидазу, выделение которой происходит замедленно. В качестве лечения рекомендуют трансплантацию костного мозга и поддерживающую симптоматическую терапию. Прогноз неблагоприятный.
Если больные доживают до 20-летнего возраста, могут появиться множественные инфильтраты в органах, которые состоят из незрелых лимфоидных клеток.
Характерными клиническими симптомами являются:
– альбинизм;
– обесцвечивание радужки глаза;
– гепатоспленомегалия;
– увеличение периферических лимфоузлов;
– рецидивирующие вирусные и кишечные инфекции;
– периферические нейропатии.
Наиболее характерным является повышенная чувствительность к гнойным инфекциям, гипопигментации кожи, волос, радужной оболочки глаз.
Поражения желудочно-кишечного тракта начинаются уже в период новорожденности, отмечается повышенная чувствительность кожи больных к солнечному свету.
Дефект пигментации связан с нарушением распределения клеток, несущих меланин коже и волосам больных, а также с недостаточной деструкцией меланосом, которые обычно располагаются аномально. В связи с этим цвет кожи варьируется от кремового до серого, цвет волос – блондин или брюнет с серым оттенком.
Со стороны крови определяется нейтропения, тромбоцитопения, анемия.
В лейкоцитах обнаруживаются гигантские включения, дающие положительную реакцию на пероксидазу; в цитоплазме нейтрофилов, моноцитов, лимфоцитов – гиганты-гранулы, содержащие миелопероксидазу, выделение которой происходит замедленно. В качестве лечения рекомендуют трансплантацию костного мозга и поддерживающую симптоматическую терапию. Прогноз неблагоприятный.
Если больные доживают до 20-летнего возраста, могут появиться множественные инфильтраты в органах, которые состоят из незрелых лимфоидных клеток.
Синдром Гуда (тимома)
Характерными клиническими симптомами являются гипертрофия тимуса за счет эпителиальных веретенообразных клеток стромы, недостаток плазматических клеток. Это проявляется низким содержанием циркулирующих лимфоцитов и всех классов иммуноглобулинов. Для данного синдрома характерна повышенная заболеваемость бактериальными, вирусными и грибковыми инфекциями, эозинопенией, эритробластемией, апластической анемией. При данном заболевании проводят удаление тимомы с последующей, симптоматической терапией возникших осложнений.
Хроническая гранулематозная болезнь
Заболевание наследуется Х-хромосомой, в редких случаях по аутосомно-рецессивному типу, возможно сцепление с полом. Диагностируется в первые 2 года жизни ребенка.
Основные симптомы:
– тяжелые инфекции кожи;
– сплено– и гепатомегалия;
– заболевания легких;
– поражение лимфоузлов;
– поражение костей и др.
Для заболевания характерны многочисленные рецидивирующие инфекции, которые возникают в ранние периоды жизни. Наиболее часто поражается кожа: сначала возникают небольшие абсцессы, быстро проникающие в подлежащие ткани и не поддающиеся лечению. Поражаются лимфатические узлы, особенно шейные, которые часто абсцедируют; могут появиться шейные свищи. Со стороны легких может диагностироваться пневмония. Возможны воспалительные процессы в пищеводе, печени и средостении.
При исследовании костного мозга обнаруживается задержка созревания миелоидных элементов. Лимфоузлы не содержат зародышевых центров, хотя содержание лимфоидных клеток и плазмоцитов в периферической крови повышено. Вилочковая железа гипоплазированная, но структура ее нормальная. При клиническом исследовании крови выражен лейкоцитоз со сдвигом влево, увеличение СОЭ, анемия. Иммуноглобулины в норме или определяется гипергаммаглобулинемия.
Прогноз хронической гранулематозной болезни неблагоприятный. Большая часть больных погибает в дошкольном возрасте, после 10 лет – доброкачественное течение.
Специфическое лечение данного заболевания не разработано. Рекомендуется применение гамма-интерферона, антимикробная терапия, симптоматическое лечение.
Основные симптомы:
– тяжелые инфекции кожи;
– сплено– и гепатомегалия;
– заболевания легких;
– поражение лимфоузлов;
– поражение костей и др.
Для заболевания характерны многочисленные рецидивирующие инфекции, которые возникают в ранние периоды жизни. Наиболее часто поражается кожа: сначала возникают небольшие абсцессы, быстро проникающие в подлежащие ткани и не поддающиеся лечению. Поражаются лимфатические узлы, особенно шейные, которые часто абсцедируют; могут появиться шейные свищи. Со стороны легких может диагностироваться пневмония. Возможны воспалительные процессы в пищеводе, печени и средостении.
При исследовании костного мозга обнаруживается задержка созревания миелоидных элементов. Лимфоузлы не содержат зародышевых центров, хотя содержание лимфоидных клеток и плазмоцитов в периферической крови повышено. Вилочковая железа гипоплазированная, но структура ее нормальная. При клиническом исследовании крови выражен лейкоцитоз со сдвигом влево, увеличение СОЭ, анемия. Иммуноглобулины в норме или определяется гипергаммаглобулинемия.
Прогноз хронической гранулематозной болезни неблагоприятный. Большая часть больных погибает в дошкольном возрасте, после 10 лет – доброкачественное течение.
Специфическое лечение данного заболевания не разработано. Рекомендуется применение гамма-интерферона, антимикробная терапия, симптоматическое лечение.
Наследственный ангионевротический отек
Заболевание обычно носит аутосомно-доминантный тип наследования. Основными провоцирующими факторами являются травмы, стресс, переохлаждение. После их воздействия могут возникнуть отеки слизистой оболочки верхних дыхательных путей, конечностей. Отеки не сопровождаются зудом. При обструкции дыхательных путей вероятно возникновение асфиксии. Иногда возникает отек кишечной стенки, которая сопровождается болями в животе, рвотой, поносом. При данной патологии выявляется дефицит ингибитора первого компонента комплемента (С1), что приводит к активации С2 и выделению вазоактивного вещества, вызывающего отеки.
В качестве лечебных мероприятий назначают введение ингибитора С1 – гепарина, аминокапроновой кислоты, регулярный прием даназола (50–600 мг/сутки). При асфиксии, связанной с отеком гортани, производится трахеостомия или трахеотомия.
В качестве лечебных мероприятий назначают введение ингибитора С1 – гепарина, аминокапроновой кислоты, регулярный прием даназола (50–600 мг/сутки). При асфиксии, связанной с отеком гортани, производится трахеостомия или трахеотомия.
Глава 4
Основные принципы диагностики и лечения первичных иммунодефицитов
Диагностика
При сборе анамнеза особое внимание обращают на следующие показатели и данные:
1. Изучение родословной ребенка с выявлением в данной семье случаев смерти детей раннего возраста от воспалительных заболеваний.
2. Выявление данных о прививочных реакциях, хронических, необычно текущих инфекциях, паразитарных и грибковых заболеваниях.
3. Выявление имеющихся среди родственников больного аллергических, аутоиммунных, опухолевидных процессов, патологии, связанной с полом, и др.
4. Развитие определенных синдромов в виде отставания в физическом развитии, кожных заболеваний с некротическими явлениями и др.
При оценке иммунной системы учитывают:
1) наличие дисплазии тимуса у детей;
2) отсутствие воспаления регионарных узлов при наличии воспалительных процессов в органах и системах;
3) наличие гипертрофии или гипоплазии миндалин, увеличение или отсутствие увеличения лимфоузлов при рецидивирующих заболеваниях.
Оцениваются также общеклинические лабораторные исследования:
1) выявление при производстве общего анализа крови гемолитической или гипопластической анемии, тромбоцитопении, лимфопении, отсутствие плазматических клеток при острой инфекции;
2) выявление при протеинограмме гипопротеинемии, гиперальбуминемии, низкого уровня B– и Г-глобулинов.
Положительные клинические симптомы для ряда наследственных заболеваний:
1) атаксия и бульбарные телеангиэктазии (синдром Луи – Бар);
2) пороки развития магистральных сосудов, судороги при синдроме Ди Джорджи.
1. Изучение родословной ребенка с выявлением в данной семье случаев смерти детей раннего возраста от воспалительных заболеваний.
2. Выявление данных о прививочных реакциях, хронических, необычно текущих инфекциях, паразитарных и грибковых заболеваниях.
3. Выявление имеющихся среди родственников больного аллергических, аутоиммунных, опухолевидных процессов, патологии, связанной с полом, и др.
4. Развитие определенных синдромов в виде отставания в физическом развитии, кожных заболеваний с некротическими явлениями и др.
При оценке иммунной системы учитывают:
1) наличие дисплазии тимуса у детей;
2) отсутствие воспаления регионарных узлов при наличии воспалительных процессов в органах и системах;
3) наличие гипертрофии или гипоплазии миндалин, увеличение или отсутствие увеличения лимфоузлов при рецидивирующих заболеваниях.
Оцениваются также общеклинические лабораторные исследования:
1) выявление при производстве общего анализа крови гемолитической или гипопластической анемии, тромбоцитопении, лимфопении, отсутствие плазматических клеток при острой инфекции;
2) выявление при протеинограмме гипопротеинемии, гиперальбуминемии, низкого уровня B– и Г-глобулинов.
Положительные клинические симптомы для ряда наследственных заболеваний:
1) атаксия и бульбарные телеангиэктазии (синдром Луи – Бар);
2) пороки развития магистральных сосудов, судороги при синдроме Ди Джорджи.
Лечение
Основные принципы лечения первичных иммунодефицитов:
– госпитализация с проведением углубленных иммунологических и молекулярных исследований;
– адекватная заместительная иммунотерапия;
– трансплантация костного мозга по показаниям.
Примерная схема лечения первичных иммунодефицитов:
1) обеспечение антибактериальной и грибковой терапии с обеспечением контроля над инфекцией;
2) применение иммунотерапии с содержанием антител:
– нативная плазма;
– иммуноглобулины для энтерального введения в виде комплексного иммуноглобулинового препарата для приема внутрь (с содержанием JgG – 50 %, JgM – 25 %, JgA – 25 %); иммуноглобулин для внутримышечного введения (ИГВМ), иммуноглобулин для внутривенного введения (ИГВВ): интроглобин Ф, интроглобин человеческий, вигам-ликвид, витам-С, октам, сандоглобулин, пептаглобин и др.
Дозировки определяются в соответствии с имеющимися инструкциями.
Для заместительной терапии применяются и другие средства. При дефиците аденозиндезаминазы производятся инъекции полиэтиленгликоля. При дефиците ингибитора С1 вводится рекомбинатный С1-ING.
В целях активации Т– и В-лимфоцитов применяются средства для лечения вторичных иммунодефицитных состояний.
Эффективна пересадка костного мозга, особенно при комбинированных иммунодефицитах. Американскими и европейскими специалистами в области генной терапии активно внедряются операции по пересадке аденозиндезаминазы, что в дальнейшем способно кардинально изменить подход к лечению заболеваний первичных иммунодефицитных состояний.
– госпитализация с проведением углубленных иммунологических и молекулярных исследований;
– адекватная заместительная иммунотерапия;
– трансплантация костного мозга по показаниям.
Примерная схема лечения первичных иммунодефицитов:
1) обеспечение антибактериальной и грибковой терапии с обеспечением контроля над инфекцией;
2) применение иммунотерапии с содержанием антител:
– нативная плазма;
– иммуноглобулины для энтерального введения в виде комплексного иммуноглобулинового препарата для приема внутрь (с содержанием JgG – 50 %, JgM – 25 %, JgA – 25 %); иммуноглобулин для внутримышечного введения (ИГВМ), иммуноглобулин для внутривенного введения (ИГВВ): интроглобин Ф, интроглобин человеческий, вигам-ликвид, витам-С, октам, сандоглобулин, пептаглобин и др.
Дозировки определяются в соответствии с имеющимися инструкциями.
Для заместительной терапии применяются и другие средства. При дефиците аденозиндезаминазы производятся инъекции полиэтиленгликоля. При дефиците ингибитора С1 вводится рекомбинатный С1-ING.
В целях активации Т– и В-лимфоцитов применяются средства для лечения вторичных иммунодефицитных состояний.
Эффективна пересадка костного мозга, особенно при комбинированных иммунодефицитах. Американскими и европейскими специалистами в области генной терапии активно внедряются операции по пересадке аденозиндезаминазы, что в дальнейшем способно кардинально изменить подход к лечению заболеваний первичных иммунодефицитных состояний.
Глава 5
Вторичные иммунодефицитные состояния
Вторичные иммунодефицитные состояния не являются генетическими дефектами и характеризуются нарушениями гуморального и клеточного иммунитета, снижением активности цитотоксических лимфоцитов и макрофагов, нарушением синтеза компонентов комплемента.
Вторичные иммунодефицитные состояния – это нарушения иммунной системы, которые развиваются у детей в постнеонатальном периоде, а также у взрослых в результате самых разнообразных причин:
– хронических заболеваний;
– дефектов питания;
– воздействия некоторых лекарственных препаратов;
– возрастной атрофии вилочковой железы;
– некачественной питьевой воды;
– чрезмерных физических нагрузок;
– воздействия радиации;
– при множественных обширных травмах;
– кортикостероидной терапии;
– после вирусных и бактериальных инфекций;
– проводимой химиотерапии;
– хирургических операций;
– спленэктомиях;
– опухолях.
Вторичные иммунодефициты представляют собой изменения в системе иммунитета, развившиеся в результате различных видов патологии и внешних воздействий.
Следствием этого являются возникновение недостаточного синтеза иммуноглобулинов, блокирование образования Т-лимфоцитов и нарушение их взаимодействия с В-лимфоцитами. Помимо этого, нередко отмечается снижение факторов неспецифической защиты.
Проявлениями вторичной иммунологической недостаточности являются:
– инфекции, трудно поддающиеся лечению;
– хронические очаги инфекции;
– длительное увеличение лимфатических узлов;
– увеличение селезенки;
– затяжные лихорадочные состояния.
Клинические проявления могут быть выражены поражениями лор-органов, желудочно-кишечного тракта, бронхолегочной системы, кожи, конъюнктивы глаз, мочеполового тракта, костной системы, септическим состоянием.
При диагностике иммунодефицитных состояний обращается внимание на анамнестические данные (личный и семейный анамнез), бытовые условия больных, профессиональный вред, проводятся консультации узких специалистов.
Особенно информативными являются дополнительные исследования:
– микробиологический анализ очагов хронической инфекции;
– клинический анализ крови с подсчетом лимфоцитов;
– проведение иммунограммы с показателем уровня иммуноглобулинов, числа Т– и В-лимфоцитов, их функциональной активности, субпопуляций Т-лимфоцитов и факторов защиты комплемента.
Вторичные иммунодефицитные состояния – это нарушения иммунной системы, которые развиваются у детей в постнеонатальном периоде, а также у взрослых в результате самых разнообразных причин:
– хронических заболеваний;
– дефектов питания;
– воздействия некоторых лекарственных препаратов;
– возрастной атрофии вилочковой железы;
– некачественной питьевой воды;
– чрезмерных физических нагрузок;
– воздействия радиации;
– при множественных обширных травмах;
– кортикостероидной терапии;
– после вирусных и бактериальных инфекций;
– проводимой химиотерапии;
– хирургических операций;
– спленэктомиях;
– опухолях.
Вторичные иммунодефициты представляют собой изменения в системе иммунитета, развившиеся в результате различных видов патологии и внешних воздействий.
Следствием этого являются возникновение недостаточного синтеза иммуноглобулинов, блокирование образования Т-лимфоцитов и нарушение их взаимодействия с В-лимфоцитами. Помимо этого, нередко отмечается снижение факторов неспецифической защиты.
Проявлениями вторичной иммунологической недостаточности являются:
– инфекции, трудно поддающиеся лечению;
– хронические очаги инфекции;
– длительное увеличение лимфатических узлов;
– увеличение селезенки;
– затяжные лихорадочные состояния.
Клинические проявления могут быть выражены поражениями лор-органов, желудочно-кишечного тракта, бронхолегочной системы, кожи, конъюнктивы глаз, мочеполового тракта, костной системы, септическим состоянием.
При диагностике иммунодефицитных состояний обращается внимание на анамнестические данные (личный и семейный анамнез), бытовые условия больных, профессиональный вред, проводятся консультации узких специалистов.
Особенно информативными являются дополнительные исследования:
– микробиологический анализ очагов хронической инфекции;
– клинический анализ крови с подсчетом лимфоцитов;
– проведение иммунограммы с показателем уровня иммуноглобулинов, числа Т– и В-лимфоцитов, их функциональной активности, субпопуляций Т-лимфоцитов и факторов защиты комплемента.
Лечение вторичных иммунодефицитов
В зависимости от причины заболевания и состояния больного назначается комплекс терапевтических мероприятий с учетом фоновых заболеваний. Из медикаментозных средств назначают витамины, адаптогены (эхинацея, элеутерококк и др.). Из лекарственных препаратов назначают курсами (2–3 раза в год):
– иммунокорректоры бактериального происхождения (рибомунил, бронхомунал);
– интерфероны (мефенамовая кислота и др.);
– вытяжки тимуса (тималин, тимоген);
– иммуномодулятор местного действия ИРС-19.
Обычно препараты назначаются в зависимости от клинических проявлений под контролем иммунологического обследования.
– иммунокорректоры бактериального происхождения (рибомунил, бронхомунал);
– интерфероны (мефенамовая кислота и др.);
– вытяжки тимуса (тималин, тимоген);
– иммуномодулятор местного действия ИРС-19.
Обычно препараты назначаются в зависимости от клинических проявлений под контролем иммунологического обследования.
Глава 6
Методы общей диагностики заболеваний иммунной системы
Необходимость иммунологического обследования возникает в случае подозрения на первичную или вторичную иммунологическую недостаточность.
Обследование назначается при наличии у больного хронических бактериальных и вирусных инфекций различных органов; при отсутствии эффекта от проводимого лечения; в случае выявления иммунологических заболеваний при обследовании иммунологического статуса; при трансплантации органов и тканей. В табл. 3 указаны клинические показания для проведения лабораторных иммунологических исследований.
Таблица 3. Клинические показания к постановке лабораторных иммунологических тестов
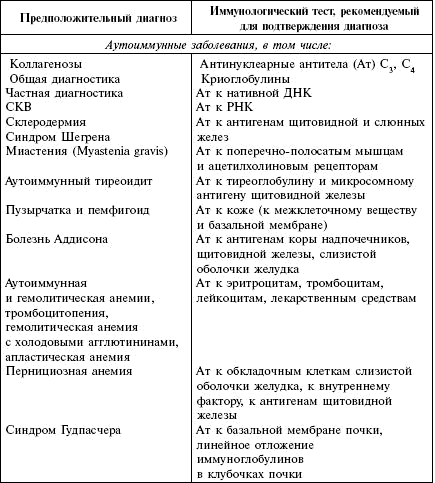



Обследование назначается при наличии у больного хронических бактериальных и вирусных инфекций различных органов; при отсутствии эффекта от проводимого лечения; в случае выявления иммунологических заболеваний при обследовании иммунологического статуса; при трансплантации органов и тканей. В табл. 3 указаны клинические показания для проведения лабораторных иммунологических исследований.
Таблица 3. Клинические показания к постановке лабораторных иммунологических тестов
Правила подготовки пациентов к обследованию, взятие и условия хранения материала для проведения исследования
Для исследования используется венозная, капиллярная кровь, слюна, желчь и другие биологические жидкости. Забор крови производится утром натощак путем пункции локтевой вены в сухую чистую пробирку. У новорожденных применяют взятие крови из пуповины. У детей раннего возраста кровь берут из височной, лобной или яремной вены. Для исследования плазмы и форменных элементов крови используются мерные пробирки, в которые вносится антикоагулянт.
Срок хранения крови от момента забора до проведения анализа составляет 2 часа (при комнатной температуре), сыворотки – 48 часов (в холодильнике). Плазму можно хранить более длительно при температуре –20 °C.
Для исследования плазмы используется также капиллярная кровь, которую берут из указательного или безымянного пальца в объеме 0,5 мл в центрифужную пробирку. Плазму получают путем центрифугирования капиллярной крови. Осадок используется для приготовления клеточной суспензии.
Перечень общих требований для исследования и транспортировки материала:
– забор крови производится утром натощак;
– материал для исследования необходимо брать в достаточном объеме;
– материал немедленно должен быть доставлен в лабораторию в специальных контейнерах. Если это невозможно, материал хранят в холодильнике в течение 48 часов;
– материал должен быть промаркирован и иметь сопроводительные документы.
В настоящее время существуют различны методы, обеспечивающие количественную и функциональную оценку иммунной системы.
Срок хранения крови от момента забора до проведения анализа составляет 2 часа (при комнатной температуре), сыворотки – 48 часов (в холодильнике). Плазму можно хранить более длительно при температуре –20 °C.
Для исследования плазмы используется также капиллярная кровь, которую берут из указательного или безымянного пальца в объеме 0,5 мл в центрифужную пробирку. Плазму получают путем центрифугирования капиллярной крови. Осадок используется для приготовления клеточной суспензии.
Перечень общих требований для исследования и транспортировки материала:
– забор крови производится утром натощак;
– материал для исследования необходимо брать в достаточном объеме;
– материал немедленно должен быть доставлен в лабораторию в специальных контейнерах. Если это невозможно, материал хранят в холодильнике в течение 48 часов;
– материал должен быть промаркирован и иметь сопроводительные документы.
В настоящее время существуют различны методы, обеспечивающие количественную и функциональную оценку иммунной системы.
Некоторые методы исследования иммунного статуса
Количественная оценка содержания иммуноглобулинов (JgA, JgM, JgG) применяется для диагностики иммунной недостаточности и контроля за иммуномодулирующей терапией.
В случае первичных иммунодефицитов определяется снижение концентрации всех иммуноглобулинов или уровень JgA и JgG при нормальных показателях JgM (врожденная гипоглобулинемия) либо снижение JgA при селективной недостаточности.
Гипергаммаглобулинемия определяется при хронических инфекционных процессах, а также системных заболеваниях соединительной ткани. Гипергаммаглобулинемия не является специфическим симптомом и встречается при многих заболеваниях: миеломной болезни, макроглобулинемии, амилоидозе и других заболеваниях.
В случае первичных иммунодефицитов определяется снижение концентрации всех иммуноглобулинов или уровень JgA и JgG при нормальных показателях JgM (врожденная гипоглобулинемия) либо снижение JgA при селективной недостаточности.
Гипергаммаглобулинемия определяется при хронических инфекционных процессах, а также системных заболеваниях соединительной ткани. Гипергаммаглобулинемия не является специфическим симптомом и встречается при многих заболеваниях: миеломной болезни, макроглобулинемии, амилоидозе и других заболеваниях.