Свет с колебаниями только в одной плоскости был назван в 1808 г. французским физиком Этьеном Луи Малюсом (1775—1812) поляризованным светом. В то время волновая теория еще не завоевала признание, и Малюс полагал, что свет состоит из частиц с северным и южным полюсами и что в поляризованном свете все полюсы ориентированы в одном направлении. Эта теория вскоре была отвергнута, но название, данное Малюсом, осталось и используется до сих пор.
Первоначально свойства и поведение поляризованного света интересовали исключительно физиков. Однако в 1815 г, французский физик Жан Батист Био (1774—1862) показал, что при прохождении поляризованного света через некоторые кристаллы происходит поворот плоскости колебаний ( плоскости поляризации) световых волн. В одних случаях она поворачивается по часовой стрелке ( правое вращение), в других — против часовой стрелки ( левое вращение). К числу кристаллов, обладающих указанным свойством,— оптической активностью, относятся и кристаллы ряда органических соединений. Более того, некоторые из этих органических соединений, например различные сахара, оптически активны и в растворах.
Со временем выяснилось, что некоторые соединения отличаются друг от друга только своими оптическими свойствами. Одно из таких одинаковых по всем другим свойствам соединений вращает плоскость поляризации поляризованного света по часовой стрелке, другое — против часовой стрелки. Обычно имеется еще и третье соединение, которое вообще не вызывает вращения плоскости поляризации поляризованного света (оптически неактивно). Примером изомерных веществ, различающихся по оптической активности, могут служить открытые Берцелиусом (см. гл. 6) виноградная и винная кислоты. Виноградная кислота оптически неактивна, а винная кислота обладает в растворе правым вращением. Позднее была открыта винная кислота, обладавшая в растворе в тех же условиях равным по величине, но противоположным, левым вращением.
Эти две формы винных кислот — природная правовращающая и не встречающаяся в природе левовращающая винная кислота — пример оптических изомеров.
Объяснить причину возникновения изомерии только с помощью структурных формул Кекуле невозможно. Первый шаг в этом направлении был сделан в 1848 г. французским химиком Луи Пастером (1822—1895). Кристаллизуя из водного раствора винограднокис-лый натрий-аммоний при комнатной температуре, Пастер обнаружил, что образованные в этих условиях кристаллы асимметричны. Причем наблюдаются две формы кристаллов: правая и левая (при одинаковой ориентации кристаллов небольшая характерная грань у одних кристаллов находилась слева, а у других — справа). Пастер сумел под увеличительным стеклом при помощи пинцета тщательно разделить оба типа кристаллов. Свойства растворов этих кристаллов оказались полностью идентичными, исключение составляла только их оптическая активность — растворы обладали противоположным вращением. Превратив кристаллы, обладающие в растворе правым вращением, в кислоту, Пастер обнаружил, что получил известную ранее природную правовращающую винную кислоту, из кристаллов другого типа получался ее оптический изомер — ранее не известная левовращающая винная кислота. Отсюда Пастер сделал вывод, что в кристаллах виноградной кислоты содержится равное количество молекул право- и левовращающих винных кислот и именно поэтому виноградная кислота оптически неактивна. Соединения, подобные виноградной кислоте, стали называть рацемическими(от латинского названия виноградной кислоты).
Результаты этих опытов убедительно свидетельствовали о том, что оптическая активность связана с асимметрией. Однако асимметрия наблюдалась у кристаллов, а многие вещества проявляли оптическую активность как в кристаллическом состоянии, так и в растворах. При растворении веществ происходит разрушение упорядоченной упаковки молекул в кристаллах, и в растворе вещества находятся в виде отдельных беспорядочно перемещающихся молекул. Если оптическая активность обусловлена асимметрией, то асимметрична должна быть и сама структура молекул.
Из структурных формул не следует, что возможно существование асимметричных молекул, однако это не позволяет говорить об отсутствии связи между асимметрией и оптической активностью. Структурные формулы записываются на плоской поверхности доски или листа бумаги, но едва ли органические молекулы в действительности являются двумерными.
Несомненно, молекулы трехмерны и образующие их атомы в действительности размещаются в трех измерениях. Расположив атомы таким образом, легко выявить ту самую асимметрию молекулы, которая обусловливает ее оптическую активность. Однако как представить себе, что молекула трехмерна?
Атомов никто никогда не видел, и само их существование могло казаться удобной выдумкой, используемой для объяснения химических реакций. Как же располагать в пространстве то, что, возможно, и не существует?
Следующий шаг мог сделать только молодой человек, еще не обремененный той мудрой осторожностью, которая приходит лишь с годами.
Молекулы в трех измерениях
Глава 8 Периодическая таблица
Приведение элементов в порядок
Первоначально свойства и поведение поляризованного света интересовали исключительно физиков. Однако в 1815 г, французский физик Жан Батист Био (1774—1862) показал, что при прохождении поляризованного света через некоторые кристаллы происходит поворот плоскости колебаний ( плоскости поляризации) световых волн. В одних случаях она поворачивается по часовой стрелке ( правое вращение), в других — против часовой стрелки ( левое вращение). К числу кристаллов, обладающих указанным свойством,— оптической активностью, относятся и кристаллы ряда органических соединений. Более того, некоторые из этих органических соединений, например различные сахара, оптически активны и в растворах.
Со временем выяснилось, что некоторые соединения отличаются друг от друга только своими оптическими свойствами. Одно из таких одинаковых по всем другим свойствам соединений вращает плоскость поляризации поляризованного света по часовой стрелке, другое — против часовой стрелки. Обычно имеется еще и третье соединение, которое вообще не вызывает вращения плоскости поляризации поляризованного света (оптически неактивно). Примером изомерных веществ, различающихся по оптической активности, могут служить открытые Берцелиусом (см. гл. 6) виноградная и винная кислоты. Виноградная кислота оптически неактивна, а винная кислота обладает в растворе правым вращением. Позднее была открыта винная кислота, обладавшая в растворе в тех же условиях равным по величине, но противоположным, левым вращением.
Эти две формы винных кислот — природная правовращающая и не встречающаяся в природе левовращающая винная кислота — пример оптических изомеров.
Объяснить причину возникновения изомерии только с помощью структурных формул Кекуле невозможно. Первый шаг в этом направлении был сделан в 1848 г. французским химиком Луи Пастером (1822—1895). Кристаллизуя из водного раствора винограднокис-лый натрий-аммоний при комнатной температуре, Пастер обнаружил, что образованные в этих условиях кристаллы асимметричны. Причем наблюдаются две формы кристаллов: правая и левая (при одинаковой ориентации кристаллов небольшая характерная грань у одних кристаллов находилась слева, а у других — справа). Пастер сумел под увеличительным стеклом при помощи пинцета тщательно разделить оба типа кристаллов. Свойства растворов этих кристаллов оказались полностью идентичными, исключение составляла только их оптическая активность — растворы обладали противоположным вращением. Превратив кристаллы, обладающие в растворе правым вращением, в кислоту, Пастер обнаружил, что получил известную ранее природную правовращающую винную кислоту, из кристаллов другого типа получался ее оптический изомер — ранее не известная левовращающая винная кислота. Отсюда Пастер сделал вывод, что в кристаллах виноградной кислоты содержится равное количество молекул право- и левовращающих винных кислот и именно поэтому виноградная кислота оптически неактивна. Соединения, подобные виноградной кислоте, стали называть рацемическими(от латинского названия виноградной кислоты).
Результаты этих опытов убедительно свидетельствовали о том, что оптическая активность связана с асимметрией. Однако асимметрия наблюдалась у кристаллов, а многие вещества проявляли оптическую активность как в кристаллическом состоянии, так и в растворах. При растворении веществ происходит разрушение упорядоченной упаковки молекул в кристаллах, и в растворе вещества находятся в виде отдельных беспорядочно перемещающихся молекул. Если оптическая активность обусловлена асимметрией, то асимметрична должна быть и сама структура молекул.
Из структурных формул не следует, что возможно существование асимметричных молекул, однако это не позволяет говорить об отсутствии связи между асимметрией и оптической активностью. Структурные формулы записываются на плоской поверхности доски или листа бумаги, но едва ли органические молекулы в действительности являются двумерными.
Несомненно, молекулы трехмерны и образующие их атомы в действительности размещаются в трех измерениях. Расположив атомы таким образом, легко выявить ту самую асимметрию молекулы, которая обусловливает ее оптическую активность. Однако как представить себе, что молекула трехмерна?
Атомов никто никогда не видел, и само их существование могло казаться удобной выдумкой, используемой для объяснения химических реакций. Как же располагать в пространстве то, что, возможно, и не существует?
Следующий шаг мог сделать только молодой человек, еще не обремененный той мудрой осторожностью, которая приходит лишь с годами.
Молекулы в трех измерениях
Таким человеком оказался молодой датский химик Якоб Гендрик Вант-Гофф (1852—1911)
[65]. В 1874 г., когда Вант-Гофф еще работал над докторской диссертацией, он выдвинул смелое предположение, согласно которому четыре связи углеродного атома направлены к четырем вершинам тетраэдра, в центре которого находится этот атом.
Представить себе это можно так: три связи атома углерода образуют треногу, а четвертая связь направлена прямо вверх. Все четыре связи при этом равноудалены друг от друга, а угол между любыми двумя соседними связями равен примерно 109° (рис. 11).
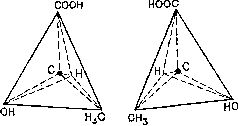
Рис. 11. Тетраэдрическое расположение связей атомов углерода допускает две конфигурации, одна из которых является зеркальным отображением другой. На рисунке показаны два возможных варианта расположения атомов в молекуле молочной кислоты.
Таким образом, четыре связи атома углерода располагаются симметрично относительно атома, и симметрия нарушается лишь в том случае, когда все четыре связи присоединяются к различным атомам или группам атомов. Поскольку присоединение может быть осуществлено двумя различными способами, полученные фигуры представляют собой зеркальные изображения друг друга. Это дает как раз тот тип асимметрии, который Пастер обнаружил в кристаллах винной кислоты.
Почти одновременно с Вант-Гоффом подобные предположения опубликовал французский химик Жозеф Ашиль Ле Бель (1847—1930). Поэтому тетраэдрическую модель атома углерода иногда называют моделью Вант-Гоффа — Ле Беля.
Гипотеза Вант-Гоффа — Ле Беля быстро завоевала признание. Этому, в частности, способствовала книга, выпущенная в 1887 г. немецким химиком Йоханнесом Адольфом Вислиценусом (1835—1902), который был широко известен в научном мире и пользовался большим авторитетом.
Соединения с асимметрическим атомом углерода (соединенным с четырьмя разными группировками) могут существовать в виде оптически активных изомеров; соединения, не имеющие таких атомов, не проявляют оптической активности.
Более того, у соединений с несколькими асимметрическими углеродами число экспериментально найденных оптически активных изомеров всегда совпадало с предсказанным на основании теории Ле Беля — Вант-Гоффа.
В конце XIX столетия утвердилось мнение, что пространственное расположение связей присуще не только атому углерода.
Немецкий химик Виктор Мейер (1848—1897) показал, что некоторые типы оптической изомерии, наблюдаемые у азотсодержащих соединений, можно объяснить, лишь допустив пространственное расположение связей азота. В 1900—1902 гг. английский химик Уильям Джексон Поуп (1870—1939) продемонстрировал, что трехмерную модель можно распространить также на атомы серы, селена и олова, а несколько позднее швейцарский химик Альфред Вернер (1866—1919) добавил к этому списку кобальт, хром, родий и ряд других металлов. (Начиная с 1891 г. Вернер занимался разработкой координационной теории, которая позволила бы объяснить свойства некоторых «необычных неорганических соединений». Согласно этой теории, кроме главных валентных сил имеются еще и силы побочной валентности. Первоначально считалось, что они резко отличаются от основных валентных сил, но впоследствии выяснилось, что существенного различия между ними не существует.
Другим важнейшим положением теории Вернера была идея о том, что группировки, связанные с атомами металла, располагаются вокруг них в пространстве в вершинах определенных многогранников (атом металла, расположенный в центре многогранника, получил название центрального атома). Теория Вернера смогла объяснить и предсказать многочисленные случаи изомерии координационных соединений, в том числе и оптической изомерии.)
С появлением трехмерной модели молекулы теория строения молекулы начала быстро развиваться. Виктор Мейер показал, что обычно группы атомов могут свободно вращаться вокруг единственной связи, соединяющей их с остальной частью молекулы, но в ряде случаев этому вращению препятствуют соседние объемные группы.
Поуп продолжил эти исследования и показал, что такая пространственно затрудненнаямолекула в целом может оказаться асимметричной и будет проявлять оптическую активность, хотя ни один из составляющих ее атомов сам по себе не является асимметрическим.
Немецкий химик Иоганн Фридрих Вильгельм Адольф фон Байер (1835—1917) использовал в 1885 г. идею трехмерного строения молекул для изображения пространственного строения циклических соединений (в виде плоских колец). Если четыре связи атомов углерода направлены к четырем углам тетраэдра, то угол между любыми двумя связями составляет 109°28'. Байер утверждал, что в любом органическом соединении атомы располагаются, как правило, так, что углы между связями атома углерода примерно соответствуют приведенному значению. Если же по какой-либо причине угол меняется, то атом оказывается в напряженном состоянии.
Если три атома углерода соединены друг с другом в цикл, то они образуют равносторонний треугольник, в котором угол между каждой парой связей равен 60°, т. е. значительно отличается от естественного угла 109°28'. По этой причине циклы из трех атомов углерода образуются с трудом, а если и образуются, то легко разрушаются.
Четыре атома углерода, согласно Байеру, образуют квадрат с углами 90°, пять атомов углерода образуют пятиугольник с углами 108°, а шесть атомов — шестиугольник с углами 120°. Вполне очевидно, что образование пятиугольника по существу не приводит к возникновению напряжений в связях атомов углерода, связи атомов в шестичленном кольце напряжены лишь в небольшой степени. Следовательно, с помощью теории напряженияБайера можно было по-видимому, объяснить, почему среди природных циклических соединений преобладают пяти- и шестичленные [66].
Однако решающей проверке теория Вант-Гоффа — Ле Беля подверглась в работах немецкого химика Эмиля Фишера (1852—1919), занимавшегося изучением простых сахаров. Ко времени начала работы Фишеру было известно, что ряд сахаров имеет одну и ту же эмпирическую формулу С 6Н 12О 6и обладает многими сходными свойствами, но различается, в частности, по оптической активности.
Фишер показал, что в молекуле каждого из этих сахаров имеются четыре асимметрических атома углерода, т. е., согласно теории Вант-Гоффа — Ле Беля, они должны иметь шестнадцать оптически активных изомеров. Эти изомеры можно расположить в виде восьми пар; в каждой такой паре изомеры вращают плоскость поляризованного света на одну и ту же величину, один по часовой стрелке, а другой — против. Фишер продолжил свою работу и установил расположение заместителей у трех асимметрических атомов углерода в молекулах ряда изомерных сахаров относительно заместителей при четвертом асимметрическом углероде, пространственное расположение которых было выбрано произвольно, поскольку в то время не существовало прямых методов его определения. (Спустя шестьдесят лет было установлено, что произвольный выбор, сделанный Фишером, оказался правильным). В результате этих работ стереохимическая теория Вант-Гоффа — Ле Беля получила наглядное и весьма впечатляющее подтверждение, что окончательно убедило химиков в ее справедливости. Предсказания теории подтвердились и при изучении ряда других соединений, в частности сахаров других типов, аминокислот и пр.
К 1900 г. трехмерная модель молекулы была принята практически всеми учеными [67].
Представить себе это можно так: три связи атома углерода образуют треногу, а четвертая связь направлена прямо вверх. Все четыре связи при этом равноудалены друг от друга, а угол между любыми двумя соседними связями равен примерно 109° (рис. 11).
Рис. 11. Тетраэдрическое расположение связей атомов углерода допускает две конфигурации, одна из которых является зеркальным отображением другой. На рисунке показаны два возможных варианта расположения атомов в молекуле молочной кислоты.
Таким образом, четыре связи атома углерода располагаются симметрично относительно атома, и симметрия нарушается лишь в том случае, когда все четыре связи присоединяются к различным атомам или группам атомов. Поскольку присоединение может быть осуществлено двумя различными способами, полученные фигуры представляют собой зеркальные изображения друг друга. Это дает как раз тот тип асимметрии, который Пастер обнаружил в кристаллах винной кислоты.
Почти одновременно с Вант-Гоффом подобные предположения опубликовал французский химик Жозеф Ашиль Ле Бель (1847—1930). Поэтому тетраэдрическую модель атома углерода иногда называют моделью Вант-Гоффа — Ле Беля.
Гипотеза Вант-Гоффа — Ле Беля быстро завоевала признание. Этому, в частности, способствовала книга, выпущенная в 1887 г. немецким химиком Йоханнесом Адольфом Вислиценусом (1835—1902), который был широко известен в научном мире и пользовался большим авторитетом.
Соединения с асимметрическим атомом углерода (соединенным с четырьмя разными группировками) могут существовать в виде оптически активных изомеров; соединения, не имеющие таких атомов, не проявляют оптической активности.
Более того, у соединений с несколькими асимметрическими углеродами число экспериментально найденных оптически активных изомеров всегда совпадало с предсказанным на основании теории Ле Беля — Вант-Гоффа.
В конце XIX столетия утвердилось мнение, что пространственное расположение связей присуще не только атому углерода.
Немецкий химик Виктор Мейер (1848—1897) показал, что некоторые типы оптической изомерии, наблюдаемые у азотсодержащих соединений, можно объяснить, лишь допустив пространственное расположение связей азота. В 1900—1902 гг. английский химик Уильям Джексон Поуп (1870—1939) продемонстрировал, что трехмерную модель можно распространить также на атомы серы, селена и олова, а несколько позднее швейцарский химик Альфред Вернер (1866—1919) добавил к этому списку кобальт, хром, родий и ряд других металлов. (Начиная с 1891 г. Вернер занимался разработкой координационной теории, которая позволила бы объяснить свойства некоторых «необычных неорганических соединений». Согласно этой теории, кроме главных валентных сил имеются еще и силы побочной валентности. Первоначально считалось, что они резко отличаются от основных валентных сил, но впоследствии выяснилось, что существенного различия между ними не существует.
Другим важнейшим положением теории Вернера была идея о том, что группировки, связанные с атомами металла, располагаются вокруг них в пространстве в вершинах определенных многогранников (атом металла, расположенный в центре многогранника, получил название центрального атома). Теория Вернера смогла объяснить и предсказать многочисленные случаи изомерии координационных соединений, в том числе и оптической изомерии.)
С появлением трехмерной модели молекулы теория строения молекулы начала быстро развиваться. Виктор Мейер показал, что обычно группы атомов могут свободно вращаться вокруг единственной связи, соединяющей их с остальной частью молекулы, но в ряде случаев этому вращению препятствуют соседние объемные группы.
Поуп продолжил эти исследования и показал, что такая пространственно затрудненнаямолекула в целом может оказаться асимметричной и будет проявлять оптическую активность, хотя ни один из составляющих ее атомов сам по себе не является асимметрическим.
Немецкий химик Иоганн Фридрих Вильгельм Адольф фон Байер (1835—1917) использовал в 1885 г. идею трехмерного строения молекул для изображения пространственного строения циклических соединений (в виде плоских колец). Если четыре связи атомов углерода направлены к четырем углам тетраэдра, то угол между любыми двумя связями составляет 109°28'. Байер утверждал, что в любом органическом соединении атомы располагаются, как правило, так, что углы между связями атома углерода примерно соответствуют приведенному значению. Если же по какой-либо причине угол меняется, то атом оказывается в напряженном состоянии.
Если три атома углерода соединены друг с другом в цикл, то они образуют равносторонний треугольник, в котором угол между каждой парой связей равен 60°, т. е. значительно отличается от естественного угла 109°28'. По этой причине циклы из трех атомов углерода образуются с трудом, а если и образуются, то легко разрушаются.
Четыре атома углерода, согласно Байеру, образуют квадрат с углами 90°, пять атомов углерода образуют пятиугольник с углами 108°, а шесть атомов — шестиугольник с углами 120°. Вполне очевидно, что образование пятиугольника по существу не приводит к возникновению напряжений в связях атомов углерода, связи атомов в шестичленном кольце напряжены лишь в небольшой степени. Следовательно, с помощью теории напряженияБайера можно было по-видимому, объяснить, почему среди природных циклических соединений преобладают пяти- и шестичленные [66].
Однако решающей проверке теория Вант-Гоффа — Ле Беля подверглась в работах немецкого химика Эмиля Фишера (1852—1919), занимавшегося изучением простых сахаров. Ко времени начала работы Фишеру было известно, что ряд сахаров имеет одну и ту же эмпирическую формулу С 6Н 12О 6и обладает многими сходными свойствами, но различается, в частности, по оптической активности.
Фишер показал, что в молекуле каждого из этих сахаров имеются четыре асимметрических атома углерода, т. е., согласно теории Вант-Гоффа — Ле Беля, они должны иметь шестнадцать оптически активных изомеров. Эти изомеры можно расположить в виде восьми пар; в каждой такой паре изомеры вращают плоскость поляризованного света на одну и ту же величину, один по часовой стрелке, а другой — против. Фишер продолжил свою работу и установил расположение заместителей у трех асимметрических атомов углерода в молекулах ряда изомерных сахаров относительно заместителей при четвертом асимметрическом углероде, пространственное расположение которых было выбрано произвольно, поскольку в то время не существовало прямых методов его определения. (Спустя шестьдесят лет было установлено, что произвольный выбор, сделанный Фишером, оказался правильным). В результате этих работ стереохимическая теория Вант-Гоффа — Ле Беля получила наглядное и весьма впечатляющее подтверждение, что окончательно убедило химиков в ее справедливости. Предсказания теории подтвердились и при изучении ряда других соединений, в частности сахаров других типов, аминокислот и пр.
К 1900 г. трехмерная модель молекулы была принята практически всеми учеными [67].
Глава 8 Периодическая таблица
Элементы в беспорядке
[68]
В истории развития органической и неорганической химии XIX столетия наблюдается любопытная параллель. В первые десятилетия число вновь открытых органических соединений, а также элементов увеличивалось ошеломляюще быстро. В третьей четверти столетия органические соединения были в определенной степени систематизированы благодаря введению структурных формул. До некоторой степени упорядочены были и элементы; отчасти этому способствовали итоги Международного химического конгресса, который состоялся в начале сентября 1860 г. в г. Карлсруэ.
Однако начнем с беспорядка, царившего в начале столетия.
Античные ученые, как известно, описали десять элементов, средневековые алхимики — четыре (см. гл. 4). В XVIII столетии были открыты такие газообразные элементы, как азот, водород, кислород и хлор, и такие металлы, как кобальт, платина, никель, марганец, вольфрам, молибден, уран, титан и хром.
В первом десятилетии XIX в. к этому списку добавилось не менее четырнадцати новых элементов Так, только Дэви выделил с помощью электролиза ни мало, ни много шесть элементов (см. гл. 4), Гей-Люссак и Тенар выделили бор, Уолластон — палладий и родий, Берцелиус открыл церий.
Английский химик Смитсон Теннант (1761—1815), у которого Уолластон работал в качестве ассистента, открыл осмий и иридий. Другой английский химик Чарльз Хэтчетт (ок. 1765—1847) выделил Колумбии (теперь официально называемый ниобием), а шведский химик Андерс Густаф Экеберг (1767—1813) открыл тантал.
Последующие десятилетия были не столь богаты открытиями, но тем не менее число элементов продолжало расти. Так, Берцелиус открыл еще четыре элемента: селен, кремний, цирконий и торий (рис. 12). Луи Никола Воклен в 1797 г. открыл бериллий.
К 1830 г. было открыто пятьдесят пять различных элементов. В теории алхимиков фигурировало всего лишь четыре элемента, и такое резкое увеличение списка элементов, которые, вдобавок, сильно отличались по свойствам, озадачило химиков. Почему элементов столько? Сколько их еще осталось открыть? Десять? Сто? Тысячу? Бесконечное число?
Заманчиво было попытаться как-то упорядочить список уже известных элементов. Может быть, при этом удастся выявить число еще неоткрытых элементов и обнаружить какую-то закономерность в изменении свойств уже открытых?
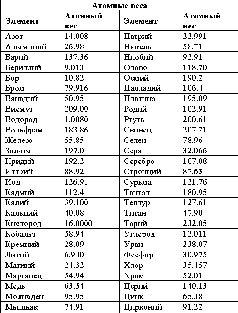
Рис. 12. Пятьдесят четыре известных во времена Берцелиуса элемента, атомные веса которых вычислены исходя из атомного веса кислорода (16.0000) (из книги «Поиски элементов»).
Первым, кому удалось уловить некоторые проблески порядка, был немецкий химик Иоганн Вольфганг Дёберейнер (1780—1849). В 1829 г., изучая свойства брома — элемента, открытого тремя годами ранее французским химиком Антуаном Жеромом Баларом (1802—1876), Дёберейнер установил, что бром по своим свойствам занимает промежуточное положение между хлором и йодом. [Йод был открыт другим французским химиком Бернаром Куртуа (1777—1838) в 1811 г.] В ряду хлор — бром — йод наблюдалось не только постепенное изменение цвета и реакционной способности, но и постепенное изменение атомного веса. Случайное совпадение?
Дёберейнер продолжил поиски и нашел еще две группы из трех элементов (он назвал их триадами), у которых также наблюдалось постепенное изменение свойств. Этими группами были кальций, стронций, барий и сера, селен, теллур. В обеих группах атомный вес среднего элемента примерно равен среднему атомных весов двух других элементов. Опять совпадение?
Дёберейнер пытался найти другие такие триады, но безуспешно. Поскольку разбить пятьдесят шесть известных элементов на триады не удалось, химики пришли к выводу, что триады Дёберейнера — явление случайное. Более того, соответствие в изменении атомных весов и химических свойств элементов в триадах Дёберейнера не произвело никакого впечатления на химиков. В первой половине XIX в. химики вообще недооценивали значение атомных весов. Атомные веса было удобно использовать при проведении разного рода расчетов, но ориентироваться на них, например, при составлении списка элементов представлялось нерезонным.
Существовали сомнения относительно целесообразности использования атомных весов в проведении расчетов. Некоторые химики не проводили четкого различия между атомным весом и молекулярным весом; некоторые путали понятия «атомный вес» и «эквивалентный вес». Так, например, эквивалентный вес кислорода равен 8 (см. гл. 5), атомный вес — 16, молекулярный вес — 32. При проведении расчетов удобнее всего пользоваться эквивалентным весом, который равен 8, почему же в таком случае для определения места кислорода в списке элементов следует использовать число 16?
Эта путаница с эквивалентным, атомным и молекулярным весами не только мешала решить вопрос о списке элементов, но и вообще отрицательно сказалась на развитии химии.
Разногласия по поводу относительных атомных весов, приписываемых различным атомам, привели к разногласиям и в отношении числа атомов отдельных элементов, входящих в данную молекулу.
Кекуле вскоре после опубликования своих предложений относительно структурных формул ясно понял, что его идея повиснет в воздухе, если химики не смогут прийти к согласию в вопросе об эмпирических формулах. Поэтому он предложил для обсуждения этого вопроса созвать конференцию ведущих химиков Европы. В результате в 1860 г. в г. Карлсруэ в Германии впервые в истории состоялась международная научная встреча химиков, получившая название «Первый международный химический конгресс».
На конгрессе присутствовало 140 делегатов, и среди них итальянский химик Станислао Канниццаро (1826—1910) [69]. Двумя годами ранее Канниццаро случайно обнаружил работу своего соотечественника Авогадро (см. гл. 5). Изучив эту работу, Канниццаро увидел, как с помощью гипотезы Авогадро можно разграничить понятия «атомный вес» и «молекулярный вес» для основных газообразных элементов и что, используя это различие, можно внести ясность в вопрос об атомных весах элементов вообще. Кроме того, он увидел, насколько важно четко отличать атомный вес от эквивалентного веса.
На конгрессе Канниццаро произнес яркую речь по этому вопросу, а затем распространил брошюру, в которой детально излагал свою точку зрения. Ему удалось убедить химиков в своей правоте, хотя произошло это не сразу и потребовало больших усилий. С этого времени в вопрос об атомных весах была внесена ясность и было по достоинству оценено значение таблицы атомных весов, составленной Берцелиусом (см. гл. 5).
Применительно к органической химии это означало, что теперь можно уже было договориться об эмпирических формулах соединений и продолжить изучение строения молекул, уточняя расположение атомов сначала плоскостное, а затем и пространственное.
В неорганической же химии теперь был принят рациональный порядок расположения элементов — в порядке увеличения их атомных весов. Как только такой список был составлен, химики смогли посмотреть на него под новым углом зрения.
Однако начнем с беспорядка, царившего в начале столетия.
Античные ученые, как известно, описали десять элементов, средневековые алхимики — четыре (см. гл. 4). В XVIII столетии были открыты такие газообразные элементы, как азот, водород, кислород и хлор, и такие металлы, как кобальт, платина, никель, марганец, вольфрам, молибден, уран, титан и хром.
В первом десятилетии XIX в. к этому списку добавилось не менее четырнадцати новых элементов Так, только Дэви выделил с помощью электролиза ни мало, ни много шесть элементов (см. гл. 4), Гей-Люссак и Тенар выделили бор, Уолластон — палладий и родий, Берцелиус открыл церий.
Английский химик Смитсон Теннант (1761—1815), у которого Уолластон работал в качестве ассистента, открыл осмий и иридий. Другой английский химик Чарльз Хэтчетт (ок. 1765—1847) выделил Колумбии (теперь официально называемый ниобием), а шведский химик Андерс Густаф Экеберг (1767—1813) открыл тантал.
Последующие десятилетия были не столь богаты открытиями, но тем не менее число элементов продолжало расти. Так, Берцелиус открыл еще четыре элемента: селен, кремний, цирконий и торий (рис. 12). Луи Никола Воклен в 1797 г. открыл бериллий.
К 1830 г. было открыто пятьдесят пять различных элементов. В теории алхимиков фигурировало всего лишь четыре элемента, и такое резкое увеличение списка элементов, которые, вдобавок, сильно отличались по свойствам, озадачило химиков. Почему элементов столько? Сколько их еще осталось открыть? Десять? Сто? Тысячу? Бесконечное число?
Заманчиво было попытаться как-то упорядочить список уже известных элементов. Может быть, при этом удастся выявить число еще неоткрытых элементов и обнаружить какую-то закономерность в изменении свойств уже открытых?
Рис. 12. Пятьдесят четыре известных во времена Берцелиуса элемента, атомные веса которых вычислены исходя из атомного веса кислорода (16.0000) (из книги «Поиски элементов»).
Первым, кому удалось уловить некоторые проблески порядка, был немецкий химик Иоганн Вольфганг Дёберейнер (1780—1849). В 1829 г., изучая свойства брома — элемента, открытого тремя годами ранее французским химиком Антуаном Жеромом Баларом (1802—1876), Дёберейнер установил, что бром по своим свойствам занимает промежуточное положение между хлором и йодом. [Йод был открыт другим французским химиком Бернаром Куртуа (1777—1838) в 1811 г.] В ряду хлор — бром — йод наблюдалось не только постепенное изменение цвета и реакционной способности, но и постепенное изменение атомного веса. Случайное совпадение?
Дёберейнер продолжил поиски и нашел еще две группы из трех элементов (он назвал их триадами), у которых также наблюдалось постепенное изменение свойств. Этими группами были кальций, стронций, барий и сера, селен, теллур. В обеих группах атомный вес среднего элемента примерно равен среднему атомных весов двух других элементов. Опять совпадение?
Дёберейнер пытался найти другие такие триады, но безуспешно. Поскольку разбить пятьдесят шесть известных элементов на триады не удалось, химики пришли к выводу, что триады Дёберейнера — явление случайное. Более того, соответствие в изменении атомных весов и химических свойств элементов в триадах Дёберейнера не произвело никакого впечатления на химиков. В первой половине XIX в. химики вообще недооценивали значение атомных весов. Атомные веса было удобно использовать при проведении разного рода расчетов, но ориентироваться на них, например, при составлении списка элементов представлялось нерезонным.
Существовали сомнения относительно целесообразности использования атомных весов в проведении расчетов. Некоторые химики не проводили четкого различия между атомным весом и молекулярным весом; некоторые путали понятия «атомный вес» и «эквивалентный вес». Так, например, эквивалентный вес кислорода равен 8 (см. гл. 5), атомный вес — 16, молекулярный вес — 32. При проведении расчетов удобнее всего пользоваться эквивалентным весом, который равен 8, почему же в таком случае для определения места кислорода в списке элементов следует использовать число 16?
Эта путаница с эквивалентным, атомным и молекулярным весами не только мешала решить вопрос о списке элементов, но и вообще отрицательно сказалась на развитии химии.
Разногласия по поводу относительных атомных весов, приписываемых различным атомам, привели к разногласиям и в отношении числа атомов отдельных элементов, входящих в данную молекулу.
Кекуле вскоре после опубликования своих предложений относительно структурных формул ясно понял, что его идея повиснет в воздухе, если химики не смогут прийти к согласию в вопросе об эмпирических формулах. Поэтому он предложил для обсуждения этого вопроса созвать конференцию ведущих химиков Европы. В результате в 1860 г. в г. Карлсруэ в Германии впервые в истории состоялась международная научная встреча химиков, получившая название «Первый международный химический конгресс».
На конгрессе присутствовало 140 делегатов, и среди них итальянский химик Станислао Канниццаро (1826—1910) [69]. Двумя годами ранее Канниццаро случайно обнаружил работу своего соотечественника Авогадро (см. гл. 5). Изучив эту работу, Канниццаро увидел, как с помощью гипотезы Авогадро можно разграничить понятия «атомный вес» и «молекулярный вес» для основных газообразных элементов и что, используя это различие, можно внести ясность в вопрос об атомных весах элементов вообще. Кроме того, он увидел, насколько важно четко отличать атомный вес от эквивалентного веса.
На конгрессе Канниццаро произнес яркую речь по этому вопросу, а затем распространил брошюру, в которой детально излагал свою точку зрения. Ему удалось убедить химиков в своей правоте, хотя произошло это не сразу и потребовало больших усилий. С этого времени в вопрос об атомных весах была внесена ясность и было по достоинству оценено значение таблицы атомных весов, составленной Берцелиусом (см. гл. 5).
Применительно к органической химии это означало, что теперь можно уже было договориться об эмпирических формулах соединений и продолжить изучение строения молекул, уточняя расположение атомов сначала плоскостное, а затем и пространственное.
В неорганической же химии теперь был принят рациональный порядок расположения элементов — в порядке увеличения их атомных весов. Как только такой список был составлен, химики смогли посмотреть на него под новым углом зрения.
Приведение элементов в порядок
В 1864 г. английский химик Джон Александер Рейна Ньюлендс (1837—1898) расположил известные элементы в порядке возрастания атомных весов. Составив такой список элементов, он обнаружил, что в полученном ряду можно выявить определенную закономерность в изменении свойств элементов (рис. 13). Когда Ньюлендс расположил элементы вертикальными столбцами по семь элементов в столбце, то выяснилось, что сходные элементы, как правило, попадают в одни и те же горизонтальные ряды. Так, калий располагается вслед за очень похожим на него натрием, селен располагается в одном ряду с похожей на него серой, кальций — рядом с похожим на него магнием и т. д. Действительно, в соответствующем ряду можно было найти каждую из трех триад Дёберейнера.

Рис. 13. «Закон октав» Ньюлендса (1864 г.).
Ньюлендс назвал открытую им закономерность законом октав, так как каждый восьмой элемент обладал свойствами, сходными с первым, девятый — со вторым и т. д.
(В музыкальной октаве семь нот, восьмая нота начинает новую октаву.) К сожалению, помимо рядов, содержащих сходные элементы, в таблице были ряды с совершенно непохожими элементами. Поэтому другие химики сочли такое совпадение случайным и отнеслись к открытию Ньюлендса как к не заслуживающему внимания факту. Ньюлендсу не удалось даже опубликовать свою работу.
Двумя годами раньше французский геолог Александр Эмиль Бегюйе де Шанкуртуа (1820—1886) также расположил элементы в порядке возрастания атомных весов и отметил их на так называемом «винтовом» графике. И в этом случае наблюдалась та же тенденция: сходные элементы попадали в вертикальные столбцы. Публикуя свое сообщение, Шанкуртуа не сопроводил его построенным им графиком, и его работа также осталась незамеченной (рис. 14).
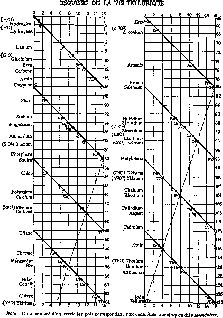
Рис. 14. «Винтовой график» Бегуйе де Шанкуртуа (1862 г.) Расположив элементы в порядке возрастания их атомных весов, ученый соединил линиями элементы с похожими свойствами.
Более удачливым оказался немецкий химик Юлиус Лотар Мейер (1830—1895). Мейер рассматривал объемы, занимаемые весовыми количествами элемента, численно равными их атомным весам. При этом выяснилось, что в каждом таком весовом количестве любого элемента содержится одно и то же число атомов. Это означало, что отношение рассматриваемых объемов различных атомов равнялось отношению объемов отдельных атомов этих элементов [70]. Поэтому указанная характеристика элемента получила название атомный объем.
Графически зависимость атомных объемов элементов от их атомных весов выражается в виде ряда волн, поднимающихся острыми пиками в точках, соответствующих щелочным металлам (натрию, калию, рубидию и цезию). Каждый спуск и подъем к пику соответствует периодув таблице элементов. В каждом периоде значения некоторых физических характеристик, помимо атомного объема, также закономерно сначала уменьшаются, а затем возрастают (рис. 15).
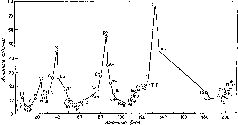
Рис. 15. График Мейера (кривая атомных объемов элементов).
Водород — элемент с наименьшим атомным весом — стоял в списке элементов первым. В то время принято было считать, что первый период включает лишь один элемент. (В современных таблицах первый период включает два элемента — водород и гелий.) Второй и третий периоды графика Мейера включали каждый по семь элементов, эти периоды дублировали октавы Ньюлендса. Однако в следующих двух периодах число элементов превышало семь. Таким образом Мейер показал, в чем ошибка Ньюлендса. Закон октав не мог строго выполняться для всего списка элементов, последние периоды должны были быть длиннее первых.
Мейер опубликовал свою работу в 1870 г. Годом раньше русский химик Дмитрий Иванович Менделеев (1834—1907) установил порядок изменения длины периодов элементов и наглядно продемонстрировал значение своего открытия [71].
Менделеев выполнял свою диссертационную работу в Германии, в Гейдельберге, как раз во время Международного химического конгресса в Карлсруэ. Он присутствовал на конгрессе и слышал речь Канниццаро, в которой тот четко изложил свою точку зрения на проблему атомного веса. Вернувшись в Россию, Менделеев приступил к изучению списка элементов и обратил внимание на периодичность изменения валентности у элементов, расположенных в порядке возрастания атомных весов: валентность водорода 1, лития 1, бериллия 2, бора 3, углерода 4, магния 2, азота 3, серы 2, фтора 1, натрия 1, алюминия 3, кремния 4, фосфора 3, кислорода 2, хлора 1 и т. д.
Основываясь на увеличении и уменьшении валентности, Менделеев разбил элементы на периоды; первый период включает только один водород, затем следуют два периода по семь элементов каждый, затем периоды, содержащие более семи элементов. Менделеев воспользовался этими данными не только для того, чтобы построить график, как это сделали Мейер и Бегюйе де Шанкуртуа, но и для того, чтобы построить таблицу, подобную таблице Ньюлендса.
Такая периодическая таблица элементовбыла яснее и нагляднее, чем график, и, кроме того, Менделеев сумел избежать ошибки Ньюлендса, настаивавшего на равенстве периодов.
Свою таблицу Менделеев опубликовал в 1869 г., т. е. раньше, чем была издана основная работа Мейера (рис. 16). Однако честь открытия Периодической системы элементов принадлежит Менделееву
Рис. 13. «Закон октав» Ньюлендса (1864 г.).
Ньюлендс назвал открытую им закономерность законом октав, так как каждый восьмой элемент обладал свойствами, сходными с первым, девятый — со вторым и т. д.
(В музыкальной октаве семь нот, восьмая нота начинает новую октаву.) К сожалению, помимо рядов, содержащих сходные элементы, в таблице были ряды с совершенно непохожими элементами. Поэтому другие химики сочли такое совпадение случайным и отнеслись к открытию Ньюлендса как к не заслуживающему внимания факту. Ньюлендсу не удалось даже опубликовать свою работу.
Двумя годами раньше французский геолог Александр Эмиль Бегюйе де Шанкуртуа (1820—1886) также расположил элементы в порядке возрастания атомных весов и отметил их на так называемом «винтовом» графике. И в этом случае наблюдалась та же тенденция: сходные элементы попадали в вертикальные столбцы. Публикуя свое сообщение, Шанкуртуа не сопроводил его построенным им графиком, и его работа также осталась незамеченной (рис. 14).
Рис. 14. «Винтовой график» Бегуйе де Шанкуртуа (1862 г.) Расположив элементы в порядке возрастания их атомных весов, ученый соединил линиями элементы с похожими свойствами.
Более удачливым оказался немецкий химик Юлиус Лотар Мейер (1830—1895). Мейер рассматривал объемы, занимаемые весовыми количествами элемента, численно равными их атомным весам. При этом выяснилось, что в каждом таком весовом количестве любого элемента содержится одно и то же число атомов. Это означало, что отношение рассматриваемых объемов различных атомов равнялось отношению объемов отдельных атомов этих элементов [70]. Поэтому указанная характеристика элемента получила название атомный объем.
Графически зависимость атомных объемов элементов от их атомных весов выражается в виде ряда волн, поднимающихся острыми пиками в точках, соответствующих щелочным металлам (натрию, калию, рубидию и цезию). Каждый спуск и подъем к пику соответствует периодув таблице элементов. В каждом периоде значения некоторых физических характеристик, помимо атомного объема, также закономерно сначала уменьшаются, а затем возрастают (рис. 15).
Рис. 15. График Мейера (кривая атомных объемов элементов).
Водород — элемент с наименьшим атомным весом — стоял в списке элементов первым. В то время принято было считать, что первый период включает лишь один элемент. (В современных таблицах первый период включает два элемента — водород и гелий.) Второй и третий периоды графика Мейера включали каждый по семь элементов, эти периоды дублировали октавы Ньюлендса. Однако в следующих двух периодах число элементов превышало семь. Таким образом Мейер показал, в чем ошибка Ньюлендса. Закон октав не мог строго выполняться для всего списка элементов, последние периоды должны были быть длиннее первых.
Мейер опубликовал свою работу в 1870 г. Годом раньше русский химик Дмитрий Иванович Менделеев (1834—1907) установил порядок изменения длины периодов элементов и наглядно продемонстрировал значение своего открытия [71].
Менделеев выполнял свою диссертационную работу в Германии, в Гейдельберге, как раз во время Международного химического конгресса в Карлсруэ. Он присутствовал на конгрессе и слышал речь Канниццаро, в которой тот четко изложил свою точку зрения на проблему атомного веса. Вернувшись в Россию, Менделеев приступил к изучению списка элементов и обратил внимание на периодичность изменения валентности у элементов, расположенных в порядке возрастания атомных весов: валентность водорода 1, лития 1, бериллия 2, бора 3, углерода 4, магния 2, азота 3, серы 2, фтора 1, натрия 1, алюминия 3, кремния 4, фосфора 3, кислорода 2, хлора 1 и т. д.
Основываясь на увеличении и уменьшении валентности, Менделеев разбил элементы на периоды; первый период включает только один водород, затем следуют два периода по семь элементов каждый, затем периоды, содержащие более семи элементов. Менделеев воспользовался этими данными не только для того, чтобы построить график, как это сделали Мейер и Бегюйе де Шанкуртуа, но и для того, чтобы построить таблицу, подобную таблице Ньюлендса.
Такая периодическая таблица элементовбыла яснее и нагляднее, чем график, и, кроме того, Менделеев сумел избежать ошибки Ньюлендса, настаивавшего на равенстве периодов.
Свою таблицу Менделеев опубликовал в 1869 г., т. е. раньше, чем была издана основная работа Мейера (рис. 16). Однако честь открытия Периодической системы элементов принадлежит Менделееву