[72]не из-за приоритета публикации, действительная причина состоит в том, как Менделеев построил свою таблицу.

Рис. 16. Страница из статьи Менделеева, опубликованной в 1869 г. в «Журнале русского химического общества». В этой статье Менделеев впервые подробно изложил основы периодической системы элементов.
Для того чтобы выполнялось требование, согласно которому в столбцах должны находиться элементы с одинаковой валентностью, Менделеев в одном или двух случаях был вынужден поместить элемент с несколько большим весом перед элементом с несколько меньшим весом. Так, теллур (атомный вес 127.6, валентность 2) пришлось поместить перед йодом (атомный вес 126.9, валентность 1), чтобы теллур попал в один столбец с элементами, валентность которых равна 2, а йод попал в один столбец с элементами, валентность которых равна 1 [73].
Поскольку этого оказалось недостаточно, Менделеев счел также необходимым оставить в своей таблице пустые места (пробелы). Причем наличие таких пробелов он объяснил не несовершенством таблицы, а тем, что соответствующие элементы пока еще не открыты.
В усовершенствованном варианте таблицы (1871 г.) существовало много пробелов, в частности не заполнены были клетки, отвечающие аналогам бора, алюминия и кремния. Менделеев был настолько уверен в своей правоте, что пришел к заключению о существовании соответствующих этим клеткам элементов и подробно описал их свойства. Он назвал их экабор, экаалюминийи экакремний(«эка» на санскрите означает «одно и то же»). Таким образом Менделеев развил идею Дёберейнера о промежуточном значении атомного веса среднего элемента в триаде; однако никто из предшественников Менделеева не рискнул предугадывать существование и свойства неоткрытых элементов.
Тем не менее часть химиков была настроена скептически, и, возможно, их недоверие еще долго не удалось бы преодолеть, если бы смелые предсказания Менделеева не подтвердились столь блестяще. Это стало возможно прежде всего благодаря применению нового физического прибора — спектроскопа.
Заполнение пробелов
Распределение новых элементов по группам
Глава 9 Физическая химия
Химическая термодинамика
Рис. 16. Страница из статьи Менделеева, опубликованной в 1869 г. в «Журнале русского химического общества». В этой статье Менделеев впервые подробно изложил основы периодической системы элементов.
Для того чтобы выполнялось требование, согласно которому в столбцах должны находиться элементы с одинаковой валентностью, Менделеев в одном или двух случаях был вынужден поместить элемент с несколько большим весом перед элементом с несколько меньшим весом. Так, теллур (атомный вес 127.6, валентность 2) пришлось поместить перед йодом (атомный вес 126.9, валентность 1), чтобы теллур попал в один столбец с элементами, валентность которых равна 2, а йод попал в один столбец с элементами, валентность которых равна 1 [73].
Поскольку этого оказалось недостаточно, Менделеев счел также необходимым оставить в своей таблице пустые места (пробелы). Причем наличие таких пробелов он объяснил не несовершенством таблицы, а тем, что соответствующие элементы пока еще не открыты.
В усовершенствованном варианте таблицы (1871 г.) существовало много пробелов, в частности не заполнены были клетки, отвечающие аналогам бора, алюминия и кремния. Менделеев был настолько уверен в своей правоте, что пришел к заключению о существовании соответствующих этим клеткам элементов и подробно описал их свойства. Он назвал их экабор, экаалюминийи экакремний(«эка» на санскрите означает «одно и то же»). Таким образом Менделеев развил идею Дёберейнера о промежуточном значении атомного веса среднего элемента в триаде; однако никто из предшественников Менделеева не рискнул предугадывать существование и свойства неоткрытых элементов.
Тем не менее часть химиков была настроена скептически, и, возможно, их недоверие еще долго не удалось бы преодолеть, если бы смелые предсказания Менделеева не подтвердились столь блестяще. Это стало возможно прежде всего благодаря применению нового физического прибора — спектроскопа.
Заполнение пробелов
В 1814 г. немецкий оптик Йозеф фон Фраунгофер (1787—1826) испытывал превосходные призмы собственного изготовления. Пропуская луч света сначала через щель, а затем через трехгранные стеклянные призмы, Фраунгофер получил солнечный спектр, пересекаемый рядом темных линий. Он насчитал около шестисот таких линий и тщательно зафиксировал их положение в спектре.
В конце 50-х годов XIX в. немецкий физик Густав Роберт Кирхгоф (1824—1887), работавший с немецким химиком Робертом Вильгельмом Бунзеном (1811—1899), показал, что эти линии содержат поразительную информацию.
В качестве источника света эти ученые пользовались изобретенной Бунзеном горелкой — той самой бунзеновской горелкой, которая известна каждому начинающему химику. Сгорающая в горелке смесь газа и воздуха дает почти бесцветное пламя с достаточно высокой температурой. Когда Кирхгоф помещал в пламя горелки крупицы различных химических веществ, оно окрашивалось в разные цвета. Свет от такого пламени, пропущенный через призму, давал не сплошную полосу, а отдельные яркие линии.
Кирхгоф показал, что для каждого элемента, разогретого в пламени горелки, характерен свой спектр. Таким образом, снимая спектр излучения химического элемента, Кирхгоф как бы снимал «отпечатки пальцев» такого элемента. Получив такую информацию, можно было решить и обратную задачу: опознать элемент, входящий в состав неизвестного вещества. Прибор, используемый для определения элементов описанным способом, получил название спектроскопа(рис. 17).
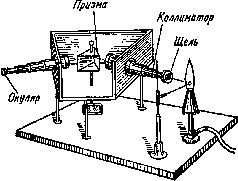
Рис. 17. Схема спектроскопа. Сравнивая спектры излучений раскаленных металлов, ученые смогли открыть новые элементы.
Сегодня мы уже знаем, что излучение света атомами обусловлено определенными явлениями, связанными с их структурой. В атомах каждого элемента эти явления протекают по-своему. Следовательно, каждый элемент испускает набор излучений только определенных длин волн.
При облучении светом элементов в парообразном состоянии наблюдается обратная картина: свет определенных длин волн не излучается, а поглощается. Более того, поскольку как поглощение, так и излучение света обусловлено одними и теми же процессами, протекающими в противоположных направлениях, то пары поглощают излучение с точно теми же длинами волн, какие наблюдаются в других условиях при испускании излучения.
Представлялось весьма вероятным, что темные линии в спектре Солнца обусловлены тем, что испускаемый раскаленной солнечной поверхностью свет поглощают газы более холодной солнечной атмосферы. Пары веществ (химических элементов), находящиеся в атмосфере Солнца, также поглощают свет определенных длин волн, и по положению возникающих темных линий в спектре можно судить, какие элементы находятся в атмосфере Солнца.
Именно спектроскоп позволил доказать, что Солнце (а также звезды и межзвездный газ) состоит из элементов, полностью идентичных земным. Этот вывод окончательно разбил утверждение Аристотеля (см. гл. 2), считавшего, что небесные тела состоят из веществ, отличающихся по своей природе от веществ, составляющих Землю.
С изобретением спектроскопа химики получили новый эффективный способ обнаружения элементов. Так, например, если в спектре раскаленного минерала содержатся линии, не принадлежащие известным элементам, то есть основания предполагать, что этот минерал содержит неизвестный элемент.
Бунзен и Кирхгоф сами продемонстрировали эффективность этого метода. В 1860 г., исследуя образец минерала, они обнаружили его в спектре линии, которые не принадлежали ни одному из известных элементов. Начав поиски нового элемента, они установили, что это щелочной металл, близкий по своим свойствам натрию и калию. Бунзен и Кирхгоф назвали открытый ими металл цезием(от латинского caesius — сине-серый), так как в спектре этого металла самой яркой была именно синяя линия. В 1861 г. эти ученые открыли еще один щелочной металл, который также назвали по цвету его спектральной линии рубидием(от латинского rubidus — темно-красный).
Новый прибор начали использовать и другие химики. Одним из них был французский химик Поль Эмиль Лекок де Буабодран (1838—1912), который в течение пятнадцати лет изучал минералы своих родных Пиренеев. В 1875 г., исследуя спектр цинковой руды, он нашел новый элемент, который назвал галлием(Галлия — древнеримское название Франции).
Спустя некоторое время Лекок де Буабодран получил такое количество этого элемента, что смог изучить его свойства. Ознакомившись с сообщением ученого, Менделеев сразу же указал, что новый элемент — это его экаалюминий. Дальнейшие исследования полностью подтвердили справедливость такого утверждения: свойства галлия оказались идентичны описанным Менделеевым свойствам экаалюминия.
Два других элемента из числа предсказанных Менделеевым были открыты старыми методами. В 1879 г. шведский химик Ларе Фредерик Нильсон (1840—1899) открыл новый элемент и назвал его скандием(в честь Скандинавии). Один из коллег Нильсона, шведский химик Пер Теодор Клеве (1840—1905). сразу же указал на сходство свойств скандия и описанного Менделеевым экабора.
Наконец, в 1886 г. немецкий химик Клеменс Александр Винклер (1838—1904), анализируя серебряную руду, установил, что на долю содержащихся в ней известных элементов приходится только 93% ее веса. Пытаясь отыскать недостающие 7%, Винклер открыл новый элемент, названный им германием(в честь Германии). Оказалось, что этот элемент идентичен экакремнию Менделеева.
Таким образом, в течение пятнадцати лет были открыты все три элемента, предсказанные Менделеевым, причем свойства всех трех элементов на удивление точно соответствовали свойствам, описанным Менделеевым. После этого в ценности и полезности периодической таблицы уже не могло быть никаких сомнений.
В конце 50-х годов XIX в. немецкий физик Густав Роберт Кирхгоф (1824—1887), работавший с немецким химиком Робертом Вильгельмом Бунзеном (1811—1899), показал, что эти линии содержат поразительную информацию.
В качестве источника света эти ученые пользовались изобретенной Бунзеном горелкой — той самой бунзеновской горелкой, которая известна каждому начинающему химику. Сгорающая в горелке смесь газа и воздуха дает почти бесцветное пламя с достаточно высокой температурой. Когда Кирхгоф помещал в пламя горелки крупицы различных химических веществ, оно окрашивалось в разные цвета. Свет от такого пламени, пропущенный через призму, давал не сплошную полосу, а отдельные яркие линии.
Кирхгоф показал, что для каждого элемента, разогретого в пламени горелки, характерен свой спектр. Таким образом, снимая спектр излучения химического элемента, Кирхгоф как бы снимал «отпечатки пальцев» такого элемента. Получив такую информацию, можно было решить и обратную задачу: опознать элемент, входящий в состав неизвестного вещества. Прибор, используемый для определения элементов описанным способом, получил название спектроскопа(рис. 17).
Рис. 17. Схема спектроскопа. Сравнивая спектры излучений раскаленных металлов, ученые смогли открыть новые элементы.
Сегодня мы уже знаем, что излучение света атомами обусловлено определенными явлениями, связанными с их структурой. В атомах каждого элемента эти явления протекают по-своему. Следовательно, каждый элемент испускает набор излучений только определенных длин волн.
При облучении светом элементов в парообразном состоянии наблюдается обратная картина: свет определенных длин волн не излучается, а поглощается. Более того, поскольку как поглощение, так и излучение света обусловлено одними и теми же процессами, протекающими в противоположных направлениях, то пары поглощают излучение с точно теми же длинами волн, какие наблюдаются в других условиях при испускании излучения.
Представлялось весьма вероятным, что темные линии в спектре Солнца обусловлены тем, что испускаемый раскаленной солнечной поверхностью свет поглощают газы более холодной солнечной атмосферы. Пары веществ (химических элементов), находящиеся в атмосфере Солнца, также поглощают свет определенных длин волн, и по положению возникающих темных линий в спектре можно судить, какие элементы находятся в атмосфере Солнца.
Именно спектроскоп позволил доказать, что Солнце (а также звезды и межзвездный газ) состоит из элементов, полностью идентичных земным. Этот вывод окончательно разбил утверждение Аристотеля (см. гл. 2), считавшего, что небесные тела состоят из веществ, отличающихся по своей природе от веществ, составляющих Землю.
С изобретением спектроскопа химики получили новый эффективный способ обнаружения элементов. Так, например, если в спектре раскаленного минерала содержатся линии, не принадлежащие известным элементам, то есть основания предполагать, что этот минерал содержит неизвестный элемент.
Бунзен и Кирхгоф сами продемонстрировали эффективность этого метода. В 1860 г., исследуя образец минерала, они обнаружили его в спектре линии, которые не принадлежали ни одному из известных элементов. Начав поиски нового элемента, они установили, что это щелочной металл, близкий по своим свойствам натрию и калию. Бунзен и Кирхгоф назвали открытый ими металл цезием(от латинского caesius — сине-серый), так как в спектре этого металла самой яркой была именно синяя линия. В 1861 г. эти ученые открыли еще один щелочной металл, который также назвали по цвету его спектральной линии рубидием(от латинского rubidus — темно-красный).
Новый прибор начали использовать и другие химики. Одним из них был французский химик Поль Эмиль Лекок де Буабодран (1838—1912), который в течение пятнадцати лет изучал минералы своих родных Пиренеев. В 1875 г., исследуя спектр цинковой руды, он нашел новый элемент, который назвал галлием(Галлия — древнеримское название Франции).
Спустя некоторое время Лекок де Буабодран получил такое количество этого элемента, что смог изучить его свойства. Ознакомившись с сообщением ученого, Менделеев сразу же указал, что новый элемент — это его экаалюминий. Дальнейшие исследования полностью подтвердили справедливость такого утверждения: свойства галлия оказались идентичны описанным Менделеевым свойствам экаалюминия.
Два других элемента из числа предсказанных Менделеевым были открыты старыми методами. В 1879 г. шведский химик Ларе Фредерик Нильсон (1840—1899) открыл новый элемент и назвал его скандием(в честь Скандинавии). Один из коллег Нильсона, шведский химик Пер Теодор Клеве (1840—1905). сразу же указал на сходство свойств скандия и описанного Менделеевым экабора.
Наконец, в 1886 г. немецкий химик Клеменс Александр Винклер (1838—1904), анализируя серебряную руду, установил, что на долю содержащихся в ней известных элементов приходится только 93% ее веса. Пытаясь отыскать недостающие 7%, Винклер открыл новый элемент, названный им германием(в честь Германии). Оказалось, что этот элемент идентичен экакремнию Менделеева.
Таким образом, в течение пятнадцати лет были открыты все три элемента, предсказанные Менделеевым, причем свойства всех трех элементов на удивление точно соответствовали свойствам, описанным Менделеевым. После этого в ценности и полезности периодической таблицы уже не могло быть никаких сомнений.
Распределение новых элементов по группам
Однако таблице Менделеева предстояла еще одна серьезная проверка — в ней должно было найтись место для других вновь открытых элементов.
Например, еще в 1794 г. финский химик Юхан Гадолин (1760—1852) предположил, что в минерале, полученном из Иттербийского карьера, расположенного вблизи Стокгольма, содержится новый оксид металла (или земля). Поскольку эта новая земля значительно отличалась от уже известных земель, например кремнезема, извести и магнезии, то ее отнесли к редким землям. Гадолин назвал открытый им оксид иттрия по названию карьера; спустя 50 лет из этого оксида был выделен в относительно чистом виде новый элемент — иттрий. Примерно в середине XIX столетия химики начали интенсивно изучать состав редкоземельных минералов. Проведенные исследования показали, что эти минералы содержат целую группу новых элементов — редкоземельных элементов. Шведский химик Карл Густав Мосандер (1797—1858) открыл, например, в конце 30-х — начале 40-х годов XIX в. четыре редкоземельных элемента: лантан, эрбий, тербийи дидим. На самом деле их было пять, поскольку спустя сорок лет в 1885 г. австрийский химик Карл Ауэр фон Вельсбах (1858—1929) обнаружил, что дидим представляет собой смесь двух элементов, которые он назвал празеодимоми неодимом. Лекок де Буабодран также открыл два редкоземельных элемента: самарийв 1879 г. и диспрозийв 1886 г. Сразу два редкоземельных элемента — гольмийи тулийописал в 1879 г. П. Т. Клеве, а в 1907 г. французский химик Жорж Урбэн (1872—1938) сообщил о новом четырнадцатом редкоземельном элементе — лютеции(Лютеция — древнее название Парижа).
Рис. 18. Современная периодическая таблица элементов.
Редкоземельные элементы обладают очень сходными химическими свойствами, их валентность равна трем. По-видимому, все эти элементы необходимо было поместить в один столбец периодической таблицы. Однако ни один из столбцов не был таким длинным, чтобы вместить четырнадцать элементов. Далее, поскольку атомные веса всех редкоземельных элементов очень близки, их следовало поместить в один горизонтальный ряд, другими словами, в один период. В принципе их можно было поместить в шестой период, если предположить, что он длиннее, чем четвертый и пятый, которые в свою очередь длиннее, чем второй и третий периоды. Однако объяснить причины сходства свойств редкоземельных элементов в то время не удалось (это было сделано лишь в 20-х годах XX в., см. гл. 13).
Другая группа вновь открытых элементов, о существовании которой во времена Менделеева химики и не подозревали, не вызвала таких затруднений; элементы этой группы прекрасно вписались в периодическую таблицу.
В 80-х годах прошлого века английский физик Джон Уильям Стратт, лорд Рэлей (1842—1919), с большой точностью определил атомные веса кислорода, водорода и азота. При этом он установил, что атомный вес азота меняется в зависимости от источника газа. Так, атомный вес азота, полученного перегонкой жидкого воздуха, немного больше, чем у азота, полученного химическим путем.
Шотландский химик Уильям Рамзай (1852—1916) заинтересовался этой проблемой и вспомнил об эксперименте Кавендиша (см. гл. 4), который еще в 1785 г. пытался связать азот воздуха с кислородом; в свое время эта работа не привлекла внимания химиков. Кавендиш установил тогда, что последний пузырек газа нельзя было заставить соединиться с кислородом ни при каких условиях. Логично было предположить, что этот последний пузырек газа мог быть и не азотом. Возможно, получаемый из воздуха азот содержит в качестве примеси другой газ, плотность которого выше, и именно поэтому полученный из воздуха азот кажется немного тяжелее, чем есть на самом деле.
В 1894 г. Рамзай повторил эксперимент Кавендиша, выделил оставшийся пузырек газа и провел его анализ новым методом, во времена Кавендиша еще неизвестным. Рамзай нагрел этот газ, изучил его спектр. В результате выяснилось, что оставшийся пузырек представляет собой новый газ, плотность которого несколько выше, чем у азота. Содержание его в атмосфере равно примерно 1% (по объему). Он химически инертен, не реагирует ни с одним другим элементом. По этой причине газ получил название аргон(от греческого ????? — инертный).
Атомный вес аргона, как выяснилось, чуть меньше 40. Это означало, что аргон должен располагаться в периодической таблице где-то возле таких элементов, как сера (атомный вес 32), хлор (атомный вес 35.5), калий (атомный вес 39) и кальций (атомный вес чуть больше 40).
Исходя только из атомного веса аргона, его следовало поместить между калием и кальцием. Однако, согласно установленному Менделеевым принципу, валентность играет более важную роль, чем атомный вес. Поскольку аргон не взаимодействует ни с одним элементом, то, следовательно, валентность его равна нулю. Куда в таком случае поместить аргон?
Валентность серы равна 2, хлора 1, калия 1 и кальция 2. Таким образом, в этой области периодической таблицы валентность меняется в следующей последовательности: 2, 1, 1, 2. Нуль в такой последовательности должен располагаться между двумя единицами; 2, 1, 0, 1, 2. Следовательно, место аргона между хлором и калием.
Однако, если принять периодическую таблицу как руководство, аргон не может существовать один. Он должен быть одним из представителей семейства инертных газов— элементов с нулевой валентностью. Столбец, занимаемый этими газами, должен располагаться между столбцами, занятыми галогенами (хлором, бромом и т. д.) и щелочными металлами (натрием, калием и т. д.); валентность и тех, и других равна единице.
Рамзай начал поиски. В 1895 г. он узнал, что в США из уранового минерала получены пробы газа — предположительно азота. Рамзай повторил эту работу и установил, что в спектре этого газа содержатся линии, которых нет ни в спектре азота, ни в спектре аргона, зато такие же линии наблюдал в солнечном спектре во время солнечного затмения 1868 г. французский астроном Пьер Жюль Сезар Жанссен (1824—1907). В то время английский астроном Джозеф Норман Локьер (1836—1920) приписал эти линии новому элементу, который он назвал гелием(от греческого ????? — Солнце).
В свое время химики почти не обратили внимания на это сообщение: новый элемент был открыт на Солнце, да еще довольно новым, не вполне завоевавшим доверие методом. Однако работа Рамзая показала, что тот же самый элемент существует и на Земле. Рамзай сохранил за элементом название, данное ему Локьером. Так был открыт гелий — самый легкий из инертных газов, который стоит вслед за водородом — элементом с наименьшим атомным весом.
В 1898 г., осторожно нагревая жидкий воздух в поиске инертных газов, которые, как предполагал Рамзай, будут испаряться первыми, он обнаружил три новых газа. Рамзай назвал их неон(новый), криптон(скрытый) и ксенон(чуждый).
Сначала считалось, что инертные газы могут представлять интерес только как объект научного исследования и никакого практического применения они не найдут. Однако в своих исследованиях, начатых им в 1910 г., французский химик Жорж Клод (1870—1960) показал, что электрический ток, пропускаемый через некоторые газы, подобные неону, вызывает мягкое окрашенное свечение.
Практическое применение этого свойства хорошо известно: таким газом можно заполнять трубки, изогнутые в виде букв, слов, фигур и т. п., и уже в 40-х годах нашего столетия улицы больших городов заливал неоновый свет [74].
Например, еще в 1794 г. финский химик Юхан Гадолин (1760—1852) предположил, что в минерале, полученном из Иттербийского карьера, расположенного вблизи Стокгольма, содержится новый оксид металла (или земля). Поскольку эта новая земля значительно отличалась от уже известных земель, например кремнезема, извести и магнезии, то ее отнесли к редким землям. Гадолин назвал открытый им оксид иттрия по названию карьера; спустя 50 лет из этого оксида был выделен в относительно чистом виде новый элемент — иттрий. Примерно в середине XIX столетия химики начали интенсивно изучать состав редкоземельных минералов. Проведенные исследования показали, что эти минералы содержат целую группу новых элементов — редкоземельных элементов. Шведский химик Карл Густав Мосандер (1797—1858) открыл, например, в конце 30-х — начале 40-х годов XIX в. четыре редкоземельных элемента: лантан, эрбий, тербийи дидим. На самом деле их было пять, поскольку спустя сорок лет в 1885 г. австрийский химик Карл Ауэр фон Вельсбах (1858—1929) обнаружил, что дидим представляет собой смесь двух элементов, которые он назвал празеодимоми неодимом. Лекок де Буабодран также открыл два редкоземельных элемента: самарийв 1879 г. и диспрозийв 1886 г. Сразу два редкоземельных элемента — гольмийи тулийописал в 1879 г. П. Т. Клеве, а в 1907 г. французский химик Жорж Урбэн (1872—1938) сообщил о новом четырнадцатом редкоземельном элементе — лютеции(Лютеция — древнее название Парижа).
Рис. 18. Современная периодическая таблица элементов.
Редкоземельные элементы обладают очень сходными химическими свойствами, их валентность равна трем. По-видимому, все эти элементы необходимо было поместить в один столбец периодической таблицы. Однако ни один из столбцов не был таким длинным, чтобы вместить четырнадцать элементов. Далее, поскольку атомные веса всех редкоземельных элементов очень близки, их следовало поместить в один горизонтальный ряд, другими словами, в один период. В принципе их можно было поместить в шестой период, если предположить, что он длиннее, чем четвертый и пятый, которые в свою очередь длиннее, чем второй и третий периоды. Однако объяснить причины сходства свойств редкоземельных элементов в то время не удалось (это было сделано лишь в 20-х годах XX в., см. гл. 13).
Другая группа вновь открытых элементов, о существовании которой во времена Менделеева химики и не подозревали, не вызвала таких затруднений; элементы этой группы прекрасно вписались в периодическую таблицу.
В 80-х годах прошлого века английский физик Джон Уильям Стратт, лорд Рэлей (1842—1919), с большой точностью определил атомные веса кислорода, водорода и азота. При этом он установил, что атомный вес азота меняется в зависимости от источника газа. Так, атомный вес азота, полученного перегонкой жидкого воздуха, немного больше, чем у азота, полученного химическим путем.
Шотландский химик Уильям Рамзай (1852—1916) заинтересовался этой проблемой и вспомнил об эксперименте Кавендиша (см. гл. 4), который еще в 1785 г. пытался связать азот воздуха с кислородом; в свое время эта работа не привлекла внимания химиков. Кавендиш установил тогда, что последний пузырек газа нельзя было заставить соединиться с кислородом ни при каких условиях. Логично было предположить, что этот последний пузырек газа мог быть и не азотом. Возможно, получаемый из воздуха азот содержит в качестве примеси другой газ, плотность которого выше, и именно поэтому полученный из воздуха азот кажется немного тяжелее, чем есть на самом деле.
В 1894 г. Рамзай повторил эксперимент Кавендиша, выделил оставшийся пузырек газа и провел его анализ новым методом, во времена Кавендиша еще неизвестным. Рамзай нагрел этот газ, изучил его спектр. В результате выяснилось, что оставшийся пузырек представляет собой новый газ, плотность которого несколько выше, чем у азота. Содержание его в атмосфере равно примерно 1% (по объему). Он химически инертен, не реагирует ни с одним другим элементом. По этой причине газ получил название аргон(от греческого ????? — инертный).
Атомный вес аргона, как выяснилось, чуть меньше 40. Это означало, что аргон должен располагаться в периодической таблице где-то возле таких элементов, как сера (атомный вес 32), хлор (атомный вес 35.5), калий (атомный вес 39) и кальций (атомный вес чуть больше 40).
Исходя только из атомного веса аргона, его следовало поместить между калием и кальцием. Однако, согласно установленному Менделеевым принципу, валентность играет более важную роль, чем атомный вес. Поскольку аргон не взаимодействует ни с одним элементом, то, следовательно, валентность его равна нулю. Куда в таком случае поместить аргон?
Валентность серы равна 2, хлора 1, калия 1 и кальция 2. Таким образом, в этой области периодической таблицы валентность меняется в следующей последовательности: 2, 1, 1, 2. Нуль в такой последовательности должен располагаться между двумя единицами; 2, 1, 0, 1, 2. Следовательно, место аргона между хлором и калием.
Однако, если принять периодическую таблицу как руководство, аргон не может существовать один. Он должен быть одним из представителей семейства инертных газов— элементов с нулевой валентностью. Столбец, занимаемый этими газами, должен располагаться между столбцами, занятыми галогенами (хлором, бромом и т. д.) и щелочными металлами (натрием, калием и т. д.); валентность и тех, и других равна единице.
Рамзай начал поиски. В 1895 г. он узнал, что в США из уранового минерала получены пробы газа — предположительно азота. Рамзай повторил эту работу и установил, что в спектре этого газа содержатся линии, которых нет ни в спектре азота, ни в спектре аргона, зато такие же линии наблюдал в солнечном спектре во время солнечного затмения 1868 г. французский астроном Пьер Жюль Сезар Жанссен (1824—1907). В то время английский астроном Джозеф Норман Локьер (1836—1920) приписал эти линии новому элементу, который он назвал гелием(от греческого ????? — Солнце).
В свое время химики почти не обратили внимания на это сообщение: новый элемент был открыт на Солнце, да еще довольно новым, не вполне завоевавшим доверие методом. Однако работа Рамзая показала, что тот же самый элемент существует и на Земле. Рамзай сохранил за элементом название, данное ему Локьером. Так был открыт гелий — самый легкий из инертных газов, который стоит вслед за водородом — элементом с наименьшим атомным весом.
В 1898 г., осторожно нагревая жидкий воздух в поиске инертных газов, которые, как предполагал Рамзай, будут испаряться первыми, он обнаружил три новых газа. Рамзай назвал их неон(новый), криптон(скрытый) и ксенон(чуждый).
Сначала считалось, что инертные газы могут представлять интерес только как объект научного исследования и никакого практического применения они не найдут. Однако в своих исследованиях, начатых им в 1910 г., французский химик Жорж Клод (1870—1960) показал, что электрический ток, пропускаемый через некоторые газы, подобные неону, вызывает мягкое окрашенное свечение.
Практическое применение этого свойства хорошо известно: таким газом можно заполнять трубки, изогнутые в виде букв, слов, фигур и т. п., и уже в 40-х годах нашего столетия улицы больших городов заливал неоновый свет [74].
Глава 9 Физическая химия
Теплота
В XVII и XVIII вв. мир химии и мир физики разделяла четкая граница. Химия изучала процессы, сопровождающиеся изменением молекулярной структуры, в то время как физика изучала такие процессы, которые подобными изменениями не сопровождались.
В начале XIX столетия, когда Дэви (см. гл. 5) разрабатывал классификацию молекул неорганических соединений, а Бертло (см. гл. 5) — классификацию молекул органических соединений, физики изучали потоки теплоты, другими словами — термодинамику(от греческого — движение тепла).
Выдающихся успехов в этой области достигли английский физик Джеймс Прескотт Джоуль (1818—1889) и немецкие физики Юлиус Роберт Майер (1814—1878) и Герман Людвиг Фердинанд Гельмгольц (1821—1894). К 40-м годам прошлого столетия в результате проведенных ими работ стало ясно, что в процессе перехода одной формы энергии в другую энергия не создается и не исчезает. Этот принцип получил название закона сохранения энергии, или первого начала термодинамики.
В своих работах французский физик Никола Леонар Сади Карно (1796—1832), английский физик Уильям Томсон, впоследствии лорд Кельвин (1824—1907), и немецкий физик Рудольф Джулиус Эмануэль Клаузиус (1822—1888) развили механическую теорию теплоты. Было показано, что при самопроизвольном переходе теплоты от точки с более высокой температурой к точке с более низкой температурой работа производится только в случае существенной разности температур, ибо часть теплоты неизбежно рассеивается в окружающую среду. Этот вывод можно обобщить и распространить на любой вид энергии.
В 1850 г. Клаузиус, пытаясь найти соотношение между количеством теплоты в изолированной системе и абсолютной температурой этой системы, ввел термин энтропия. Он показал, что при любых самопроизвольных изменениях энергии энтропия системы должна увеличиваться. Этот принцип был назван вторым началом термодинамики.
Естественно, что такого рода открытия не могли не повлиять на развитие химии. Ведь в конечном итоге основными источниками теплоты в XIX в. (кроме Солнца) были химические реакции: горение дерева, угля и нефти. Химикам было также известно, что теплота выделяется и при других химических реакциях, например при нейтрализации кислот основаниями, и что практически все химические реакции сопровождаются тем или иным тепловым эффектом: выделением теплоты (а иногда и света) или поглощением теплоты (а иногда и света).
В 1840 г. после опубликования работ русского химика Германа Ивановича Гесса (1802—1850) [75]граница между миром физики и химии была разрушена, и началось сотрудничество двух наук. Тщательно измерив действительное количество теплоты, выделяемой в процессе химических реакций между определенными количествами веществ, Гесс показал, что количество теплоты, получаемой (или поглощаемой) при переходе от одного вещества к другому, всегда одинаково и не зависит от того, с помощью какой химической реакции или сколькими этапами осуществляется этот переход. Благодаря этому обобщению ( закон Гесса) Гесса иногда считают основателем термохимии(теплохимии).
Исходя из закона Гесса, представлялось вполне вероятным, что закон сохранения энергии равно применим и к химическим, и к физическим процессам. И действительно, дальнейшие обобщения показали, что законы термодинамики, вероятнее всего, проявляются в химии точно так же, как и в физике.
Это направление в экспериментах и в теории привело к выводу, что определенным химическим реакциям, как и физическим процессам, присуще свойственное только им самопроизвольное направление, приводящее к увеличению энтропии. Однако энтропия представляет собой величину, трудную для непосредственного измерения, поэтому химики начали искать другой, более простой критерий.
В 60-х годах прошлого столетия Бертло, уже завоевавший известность как органик-синтетик (см. гл. 5), обратился к термохимии. Он разработал методику проведения химических реакций в замкнутых сосудах, погруженных в воду заданной температуры. Определив температуру этой воды в конце реакции, можно было установить, какое количество теплоты выделяется в ходе данной реакции.
Используя такой калориметр(от латинского calorimeter — измерение тепла), Бертло тщательно измерил количество теплоты, выделяемой в результате сотен различных химических реакций. Подобные эксперименты независимо от Бертло провел также датский химик Ханс Петер Юрген Юлиус Томсен (1826—1909).
Бертло полагал, что реакции, сопровождающиеся выделением теплоты, являются самопроизвольными, в то время как реакции, сопровождающиеся поглощением теплоты, таковыми не являются. Поскольку каждая реакция, в ходе которой выделяется теплота, должна сопровождаться, если заставить ее идти в обратном направлении, поглощением теплоты (первыми стали придерживаться такой точки зрения Лавуазье и Лаплас, см. гл. 4), то, следовательно, любая химическая реакция идет самопроизвольно только в одном направлении, и при этом она сопровождается выделением теплоты.
Например, когда водород взаимодействует с кислородом, образуя воду, реакция протекает с выделением большого количества теплоты. Эта реакция самопроизвольная, и, однажды начавшись, она быстро идет к завершению и иногда заканчивается сильным взрывом.
В то же время обратная реакция — расщепление воды на водород и кислород — требует затраты энергии (тепловой или, лучше, электрической). Расщепление молекулы воды не является самопроизвольным; в отсутствие энергии расщепление вообще не происходит, и уже начавшаяся реакция тотчас же прекратится, если подачу энергии прервать.
Но это правило Бертло, на первый взгляд представлявшееся вполне приемлемым, было ошибочным. Во-первых, не все самопроизвольные реакции протекают с выделением теплоты; некоторые реакции сопровождаются поглощением теплоты, и в ходе таких реакций температура среды, окружающей реакционную смесь, действительно понижается.
Во-вторых, существуют обратимые реакции. Так, например, вещества A и B могут самопроизвольно взаимодействовать и превращаться в вещества C и D, которые в свою очередь могут вновь самопроизвольно образовать вещества A и B. И это несмотря на то, что если какая-либо реакция сопровождается выделением теплоты, то обратная ей реакция должна сопровождаться поглощением теплоты. Например, иодид водорода разлагается на йод и водород, которые вновь могут образовывать иодид водорода:
H 2 + I 2 ? 2HI
(две стрелки, направленные в противоположные стороны, показывают, что реакция обратима).
Во времена Бертло обратимые реакции были уже известны. В 1850 г. Уильямсон первым тщательно изучил их. Основываясь на результатах проведенных им работ, Уильямсон (см. гл. 7) предложил структурные формулы эфиров. Он нашел условия, при которых смесь веществ A и B образовывала вещества C и D, а смесь веществ C и D образовывала вещества A и B. Однако и в том, и в другом случае в итоге получалась смесь веществ A, B, C и D, причем соотношение этих компонентов было определенным. Смесь при этом находилась в состоянии равновесия. Хотя состав смеси оставался скорее всего постоянным, тем не менее Уильямсон считал, что вещества A и B реагируют, образуя вещества C и D, а вещества C и D реагируют, образуя вещества A и B. Обе реакции идут непрерывно, но они нейтрализуют друг друга, создавая иллюзию покоя, тогда как в действительности смесь находится в состоянии динамического равновесия.
Работа Уильямсона ознаменовала начало изучения химической кинетики— области химии, изучающей скорости химических реакций. Уильямсон ясно показал, что самопроизвольный характер химической реакции в ряде случаев определяет не просто выделение теплоты, а нечто большее. Проводя свои многочисленные калориметрические измерения, Бертло и Томсен уже выявили это «нечто большее», но, к сожалению, вопрос остался нерешенным из-за того, что работы Томсена были опубликованы на малодоступном ученым норвежском языке.
В начале XIX столетия, когда Дэви (см. гл. 5) разрабатывал классификацию молекул неорганических соединений, а Бертло (см. гл. 5) — классификацию молекул органических соединений, физики изучали потоки теплоты, другими словами — термодинамику(от греческого — движение тепла).
Выдающихся успехов в этой области достигли английский физик Джеймс Прескотт Джоуль (1818—1889) и немецкие физики Юлиус Роберт Майер (1814—1878) и Герман Людвиг Фердинанд Гельмгольц (1821—1894). К 40-м годам прошлого столетия в результате проведенных ими работ стало ясно, что в процессе перехода одной формы энергии в другую энергия не создается и не исчезает. Этот принцип получил название закона сохранения энергии, или первого начала термодинамики.
В своих работах французский физик Никола Леонар Сади Карно (1796—1832), английский физик Уильям Томсон, впоследствии лорд Кельвин (1824—1907), и немецкий физик Рудольф Джулиус Эмануэль Клаузиус (1822—1888) развили механическую теорию теплоты. Было показано, что при самопроизвольном переходе теплоты от точки с более высокой температурой к точке с более низкой температурой работа производится только в случае существенной разности температур, ибо часть теплоты неизбежно рассеивается в окружающую среду. Этот вывод можно обобщить и распространить на любой вид энергии.
В 1850 г. Клаузиус, пытаясь найти соотношение между количеством теплоты в изолированной системе и абсолютной температурой этой системы, ввел термин энтропия. Он показал, что при любых самопроизвольных изменениях энергии энтропия системы должна увеличиваться. Этот принцип был назван вторым началом термодинамики.
Естественно, что такого рода открытия не могли не повлиять на развитие химии. Ведь в конечном итоге основными источниками теплоты в XIX в. (кроме Солнца) были химические реакции: горение дерева, угля и нефти. Химикам было также известно, что теплота выделяется и при других химических реакциях, например при нейтрализации кислот основаниями, и что практически все химические реакции сопровождаются тем или иным тепловым эффектом: выделением теплоты (а иногда и света) или поглощением теплоты (а иногда и света).
В 1840 г. после опубликования работ русского химика Германа Ивановича Гесса (1802—1850) [75]граница между миром физики и химии была разрушена, и началось сотрудничество двух наук. Тщательно измерив действительное количество теплоты, выделяемой в процессе химических реакций между определенными количествами веществ, Гесс показал, что количество теплоты, получаемой (или поглощаемой) при переходе от одного вещества к другому, всегда одинаково и не зависит от того, с помощью какой химической реакции или сколькими этапами осуществляется этот переход. Благодаря этому обобщению ( закон Гесса) Гесса иногда считают основателем термохимии(теплохимии).
Исходя из закона Гесса, представлялось вполне вероятным, что закон сохранения энергии равно применим и к химическим, и к физическим процессам. И действительно, дальнейшие обобщения показали, что законы термодинамики, вероятнее всего, проявляются в химии точно так же, как и в физике.
Это направление в экспериментах и в теории привело к выводу, что определенным химическим реакциям, как и физическим процессам, присуще свойственное только им самопроизвольное направление, приводящее к увеличению энтропии. Однако энтропия представляет собой величину, трудную для непосредственного измерения, поэтому химики начали искать другой, более простой критерий.
В 60-х годах прошлого столетия Бертло, уже завоевавший известность как органик-синтетик (см. гл. 5), обратился к термохимии. Он разработал методику проведения химических реакций в замкнутых сосудах, погруженных в воду заданной температуры. Определив температуру этой воды в конце реакции, можно было установить, какое количество теплоты выделяется в ходе данной реакции.
Используя такой калориметр(от латинского calorimeter — измерение тепла), Бертло тщательно измерил количество теплоты, выделяемой в результате сотен различных химических реакций. Подобные эксперименты независимо от Бертло провел также датский химик Ханс Петер Юрген Юлиус Томсен (1826—1909).
Бертло полагал, что реакции, сопровождающиеся выделением теплоты, являются самопроизвольными, в то время как реакции, сопровождающиеся поглощением теплоты, таковыми не являются. Поскольку каждая реакция, в ходе которой выделяется теплота, должна сопровождаться, если заставить ее идти в обратном направлении, поглощением теплоты (первыми стали придерживаться такой точки зрения Лавуазье и Лаплас, см. гл. 4), то, следовательно, любая химическая реакция идет самопроизвольно только в одном направлении, и при этом она сопровождается выделением теплоты.
Например, когда водород взаимодействует с кислородом, образуя воду, реакция протекает с выделением большого количества теплоты. Эта реакция самопроизвольная, и, однажды начавшись, она быстро идет к завершению и иногда заканчивается сильным взрывом.
В то же время обратная реакция — расщепление воды на водород и кислород — требует затраты энергии (тепловой или, лучше, электрической). Расщепление молекулы воды не является самопроизвольным; в отсутствие энергии расщепление вообще не происходит, и уже начавшаяся реакция тотчас же прекратится, если подачу энергии прервать.
Но это правило Бертло, на первый взгляд представлявшееся вполне приемлемым, было ошибочным. Во-первых, не все самопроизвольные реакции протекают с выделением теплоты; некоторые реакции сопровождаются поглощением теплоты, и в ходе таких реакций температура среды, окружающей реакционную смесь, действительно понижается.
Во-вторых, существуют обратимые реакции. Так, например, вещества A и B могут самопроизвольно взаимодействовать и превращаться в вещества C и D, которые в свою очередь могут вновь самопроизвольно образовать вещества A и B. И это несмотря на то, что если какая-либо реакция сопровождается выделением теплоты, то обратная ей реакция должна сопровождаться поглощением теплоты. Например, иодид водорода разлагается на йод и водород, которые вновь могут образовывать иодид водорода:
H 2 + I 2 ? 2HI
(две стрелки, направленные в противоположные стороны, показывают, что реакция обратима).
Во времена Бертло обратимые реакции были уже известны. В 1850 г. Уильямсон первым тщательно изучил их. Основываясь на результатах проведенных им работ, Уильямсон (см. гл. 7) предложил структурные формулы эфиров. Он нашел условия, при которых смесь веществ A и B образовывала вещества C и D, а смесь веществ C и D образовывала вещества A и B. Однако и в том, и в другом случае в итоге получалась смесь веществ A, B, C и D, причем соотношение этих компонентов было определенным. Смесь при этом находилась в состоянии равновесия. Хотя состав смеси оставался скорее всего постоянным, тем не менее Уильямсон считал, что вещества A и B реагируют, образуя вещества C и D, а вещества C и D реагируют, образуя вещества A и B. Обе реакции идут непрерывно, но они нейтрализуют друг друга, создавая иллюзию покоя, тогда как в действительности смесь находится в состоянии динамического равновесия.
Работа Уильямсона ознаменовала начало изучения химической кинетики— области химии, изучающей скорости химических реакций. Уильямсон ясно показал, что самопроизвольный характер химической реакции в ряде случаев определяет не просто выделение теплоты, а нечто большее. Проводя свои многочисленные калориметрические измерения, Бертло и Томсен уже выявили это «нечто большее», но, к сожалению, вопрос остался нерешенным из-за того, что работы Томсена были опубликованы на малодоступном ученым норвежском языке.
Химическая термодинамика
В 1863 г. норвежские химики Като Максимилиан Гульдберг (1836—1902) и Петер Вааге (1833—1900) опубликовали брошюру, в которой излагали свою точку зрения на причины, определяющие направление течения самопроизвольных реакций. Эти ученые вернулись к предположению, высказанному Бертолле (см. гл. 4) за полстолетия до этого. Бертолле считал, что направление реакции зависит от массы участвующих в ней отдельных веществ. Гульдберг и Вааге полагали, что направление реакции определяется не просто массой отдельных веществ, а скорее массой отдельных веществ, приходящейся на данный объем реагирующей смеси, другими словами —
концентрациейвеществ.
Предположим, что вещества A и B могут реагировать с образованием веществ C и D, а вещества C и D могут реагировать с образованием веществ A и B:
A + B ? C + D
Эта обратимая реакция достигает равновесия при таких условиях, когда в системе представлены все четыре вещества: A. B, C и D. Положение равновесия зависит от соотношения скоростей реакций веществ A и B (скорость 1) и веществ C и D (скорость 2).
Предположим, что скорость 1намного больше, чем скорость 2. В этом случае вещества A и B реагируют быстро, а вещества C и D — медленно, и вскоре количество веществ C и D намного превысит количество веществ A и B, и в состоянии равновесия в смеси преобладают вещества C и D. Взглянув на приведенное выше уравнение, мы скажем, что в этом случае точка равновесия сдвинута «далеко вправо».
Если же скорость 2намного выше, чем скорость 1, вещества C и D реагируют намного быстрее, чем вещества A и B, и в состоянии равновесия в смеси преобладают вещества A и B. Точка равновесия в этом случае сдвинута «далеко влево».
Предположим, что вещества A и B могут реагировать с образованием веществ C и D, а вещества C и D могут реагировать с образованием веществ A и B:
A + B ? C + D
Эта обратимая реакция достигает равновесия при таких условиях, когда в системе представлены все четыре вещества: A. B, C и D. Положение равновесия зависит от соотношения скоростей реакций веществ A и B (скорость 1) и веществ C и D (скорость 2).
Предположим, что скорость 1намного больше, чем скорость 2. В этом случае вещества A и B реагируют быстро, а вещества C и D — медленно, и вскоре количество веществ C и D намного превысит количество веществ A и B, и в состоянии равновесия в смеси преобладают вещества C и D. Взглянув на приведенное выше уравнение, мы скажем, что в этом случае точка равновесия сдвинута «далеко вправо».
Если же скорость 2намного выше, чем скорость 1, вещества C и D реагируют намного быстрее, чем вещества A и B, и в состоянии равновесия в смеси преобладают вещества A и B. Точка равновесия в этом случае сдвинута «далеко влево».