Страница:
Гиповитаминоз А. Физиологические функции витамина А связаны с процессами фоторецепции, роста, регенерации и дифференцировки эпителия и соединительной ткани, иммунологически – с гомеостазом. Гиповитаминоз А проявляется в виде трех основных патологических процессов:
1) гемералопии (куриная слепота) – резкого ухудшения зрения при недостаточном освещении, в сумерках вследствие дефицита фоточувствительного протеида родопсина, содержащего витамин А;
2) ксерофтальмии, связанной с гиперкератозом и закупоркой эпителием слезных протоков, высыханием конъюнктивы и роговой оболочки глаза;
3) усилением кератинизации, обусловленным уменьшением интенсивности восстановления эпителия и слизистых оболочек, увеличением ороговения, дистрофией и слущиванием эпителия. В секреторных железах (слюнных, поджелудочной) интенсивно идет процесс гиалинизации, возникает недостаточность их функции.
Гипервитаминоз А связан с интенсификацией обмена и развитием дистрофических процессов в печени, почках, сердце, костях. Наблюдается при передозировке витамина А или избыточном потреблении содержащих его продуктов.
Авитаминоз D. Дефицит кальциферола в организме приводит к развитию рахита. Обмен витамина D достаточно сложен, в его синтезе и трансформации принимают участие ткани кожи, печени, почек. При недостаточности витамина в пище, дефиците инсоляции или развитии патологии указанных органов возникают изменения психики ребенка (вялость, плаксивость, нарушения сна), наблюдаются потливость, задержка роста, искривления костей и задержка сроков окостенения. У взрослых возможно размягчение костной ткани (остеомаляция).
Гипервитаминоз D. При передозировке витамина D прекращается рост, утолщаются кости, рано зарастает родничок, возникает микроцефалия. Кроме того, фосфат кальция откладывается в тканях разных органов: в стенках сосудов, почках, миокарде. Кальцинаты становятся местом развития склероза. Нередко присоединяется мочекаменная и желчекаменная болезнь.
Авитаминоз С. Цинга, или скорбут, – один из первых описанных авитаминозов. При авитаминозе С нарушаются окислительно-восстановительные процессы, наблюдаются и другие изменения:
• гипофункция надпочечников (витамин С напрямую связан с образованием кортикостероидов);
• снижение активности углеводного и энергетического обмена;
• торможение синтеза белка при усилении его распада. Развиваются мощные дистрофические процессы в тканях, на слизистых оболочках; отмечаются дефицит иммунитета, нарушение образования коллагена, процессов окостенения.
Авитаминоз В1 (болезнь бери-бери). Витамин В1 в виде тиаминпирофосфата входит в состав ключевых ферментов энергетического обмена. Основное проявление авитаминоза В1 связано с накоплением в тканях молочной и пировиноградной кислот, развитием ацидоза, раздражением нервных окончаний, торможением окислительного фосфорилирования, биосинтеза нуклеиновых кислот и белков. Уменьшается масса тела, наблюдается общая дистрофия, развиваются неврологические изменения.
Гиповитаминоз В12. Цианокобаламин поступает в организм с мясными продуктами, всасывается в желудке при участии специального переносчика (фактор Кастла), участвует в обмене нуклеотидов, входит в состав гемоглобина. Дефицит витамина В12 возникает вследствие: алиментарной недостаточности витамина, дефицита поверхности всасывания или фактора Кастла, потреблении внутрикишечными гельминтами (в основном при дифиллоботриозе).
Основное проявление авитаминоза – гиперхромная, так называемая злокачественная, В12-дефицитная анемия.
Авитаминоз PP. Основные причины дефицита витамина РР (никотиновой кислоты) – алиментарная недостаточность и нарушение биосинтеза никотиновой кислоты в организме.
При авитаминозе РР снижается биосинтез адениловых нуклеотидов и тормозится окислительное фосфорилирование. В результате развивается пеллагра – заболевание, характеризующееся триадой «Д»: дерматит, функциональная диарея, деменция (прогрессирующее нарушение психики).
6.4. ПАТОЛОГИЯ УГЛЕВОДНОГО ОБМЕНА. УГЛЕВОДНЫЕ ДИСТРОФИИ
6.5. ПАТОЛОГИЯ ЛИПИДНОГО ОБМЕНА. ЛИПИДНЫЕ ДИСТРОФИИ
6.6. ДИСГИДРИИ. ОТЕКИ
6.7. НАРУШЕНИЯ ОБМЕНА ЭЛЕКТРОЛИТОВ
1) гемералопии (куриная слепота) – резкого ухудшения зрения при недостаточном освещении, в сумерках вследствие дефицита фоточувствительного протеида родопсина, содержащего витамин А;
2) ксерофтальмии, связанной с гиперкератозом и закупоркой эпителием слезных протоков, высыханием конъюнктивы и роговой оболочки глаза;
3) усилением кератинизации, обусловленным уменьшением интенсивности восстановления эпителия и слизистых оболочек, увеличением ороговения, дистрофией и слущиванием эпителия. В секреторных железах (слюнных, поджелудочной) интенсивно идет процесс гиалинизации, возникает недостаточность их функции.
Гипервитаминоз А связан с интенсификацией обмена и развитием дистрофических процессов в печени, почках, сердце, костях. Наблюдается при передозировке витамина А или избыточном потреблении содержащих его продуктов.
Авитаминоз D. Дефицит кальциферола в организме приводит к развитию рахита. Обмен витамина D достаточно сложен, в его синтезе и трансформации принимают участие ткани кожи, печени, почек. При недостаточности витамина в пище, дефиците инсоляции или развитии патологии указанных органов возникают изменения психики ребенка (вялость, плаксивость, нарушения сна), наблюдаются потливость, задержка роста, искривления костей и задержка сроков окостенения. У взрослых возможно размягчение костной ткани (остеомаляция).
Гипервитаминоз D. При передозировке витамина D прекращается рост, утолщаются кости, рано зарастает родничок, возникает микроцефалия. Кроме того, фосфат кальция откладывается в тканях разных органов: в стенках сосудов, почках, миокарде. Кальцинаты становятся местом развития склероза. Нередко присоединяется мочекаменная и желчекаменная болезнь.
Авитаминоз С. Цинга, или скорбут, – один из первых описанных авитаминозов. При авитаминозе С нарушаются окислительно-восстановительные процессы, наблюдаются и другие изменения:
• гипофункция надпочечников (витамин С напрямую связан с образованием кортикостероидов);
• снижение активности углеводного и энергетического обмена;
• торможение синтеза белка при усилении его распада. Развиваются мощные дистрофические процессы в тканях, на слизистых оболочках; отмечаются дефицит иммунитета, нарушение образования коллагена, процессов окостенения.
Авитаминоз В1 (болезнь бери-бери). Витамин В1 в виде тиаминпирофосфата входит в состав ключевых ферментов энергетического обмена. Основное проявление авитаминоза В1 связано с накоплением в тканях молочной и пировиноградной кислот, развитием ацидоза, раздражением нервных окончаний, торможением окислительного фосфорилирования, биосинтеза нуклеиновых кислот и белков. Уменьшается масса тела, наблюдается общая дистрофия, развиваются неврологические изменения.
Гиповитаминоз В12. Цианокобаламин поступает в организм с мясными продуктами, всасывается в желудке при участии специального переносчика (фактор Кастла), участвует в обмене нуклеотидов, входит в состав гемоглобина. Дефицит витамина В12 возникает вследствие: алиментарной недостаточности витамина, дефицита поверхности всасывания или фактора Кастла, потреблении внутрикишечными гельминтами (в основном при дифиллоботриозе).
Основное проявление авитаминоза – гиперхромная, так называемая злокачественная, В12-дефицитная анемия.
Авитаминоз PP. Основные причины дефицита витамина РР (никотиновой кислоты) – алиментарная недостаточность и нарушение биосинтеза никотиновой кислоты в организме.
При авитаминозе РР снижается биосинтез адениловых нуклеотидов и тормозится окислительное фосфорилирование. В результате развивается пеллагра – заболевание, характеризующееся триадой «Д»: дерматит, функциональная диарея, деменция (прогрессирующее нарушение психики).
6.4. ПАТОЛОГИЯ УГЛЕВОДНОГО ОБМЕНА. УГЛЕВОДНЫЕ ДИСТРОФИИ
Углеводы могут синтезироваться в организме из неуглеводных соединений (глюконеогенез), поэтому алиментарный дефицит углеводов существенной роли в патологии не играет. Дефицит в пище растительных полисахаридов может вызвать снижение моторики кишечника, а избыток моно– и дисахаридов (рафинированные углеводы) – усиление бродильных процессов в кишечнике. Приводим основные виды нарушений углеводного обмена.
Недостаточность расщепления и всасывания углеводов. Дефицит амилазы в кишечнике может быть связан с недостаточностью функции поджелудочной железы, закупоркой ее протока камнем или опухолью. Относительная недостаточность возникает при усилении моторики кишечника (сокращение времени на переваривание). Всасывание углеводов наблюдается при воспалительных и атрофических процессах в кишечнике, отравлениях, действии ряда лекарственных препаратов. Оно резко уменьшается при наследственном или приобретенном дефиците ферментов (сахаридаз), ответственных за конечные этапы пристеночного пищеварения углеводов и проникновение их через стенку кишки.
Нарушения обмена гликогена. В норме в печени содержится гликогена до 5 % от массы органа, что составляет суточный запас для воспроизводства энергии в аэробном окислении. Запасы гликогена имеются в скелетных мышцах, миокарде, почках. При повышенном питании эти запасы возрастают, при голодании или резком увеличении потребности в энергии гликоген расходуется. Распад гликогена (гликогенолиз) активируется при стрессе, возбуждении ЦНС любого генеза, тканевой гипоксии жизненно важных органов. Дефицит гликогена сказывается на детоксицирующей функции печени, вызывает мобилизацию жира из депо и переключение энергетики на белковый и жировой обмен с тяжелыми клеточными дистрофиями.
Недостаточность утилизации глюкозы. Такая недостаточность приводит к накоплению определенного углеводсодержащего продукта в ткани. Классический пример – гликогенозы (6 типов) и мукополисахаридозы (2 типа). Гликогеноз – это общее название болезней, для которых характерно избыточное накопление гликогена в печени при наследственном дефиците ферментов гликогенолиза. К таким болезням относятся болезнь Гирке (дефицит глюкозо-6-фосфатазы), когда гликоген накапливается в печени и почках; болезнь Помпе, при которой избыток гликогена обнаруживается в скелетных мышцах и сердце. Галактоземия – наследственное заболевание, связанное с дефицитом фермента галактозо-1-фосфатуридилтрансферазы. Накопление галактозо-1-фосфата происходит в крови, хрусталике (катаракта и слепота), печени, селезенке. Развиваются задержка роста и умственная отсталость, нарастает масса тела. Лечение состоит в полном исключении галактозы из диеты.
Нарушения межуточного обмена. Основной путь межуточного обмена углеводов направлен на подготовку субстратов аэробного окисления. При дефиците кислорода (гипоксия), а также при различных нарушениях в ферментах дыхательной цепи (например, при авитаминозе В1) происходит значительная активация анаэробного окисления с накоплением в крови и тканях пировиноградной кислоты и лактата, смещением рН в кислую сторону.
Другие нарушения межуточного обмена обусловлены изменением скорости реакций глюконеогенеза и пентозофосфатного цикла, связывающего углеводный обмен с другими видами метаболизма.
Нарушения концентрации глюкозы в крови. Гипергликемия – содержание глюкозы в кpови выше нормы (3,3–5,5 ммоль/л) (схема 6.1).
Схема 6.1. Патогенез гипергликемии у больных сахарным диабетом [Шанин В. Ю., 1998]
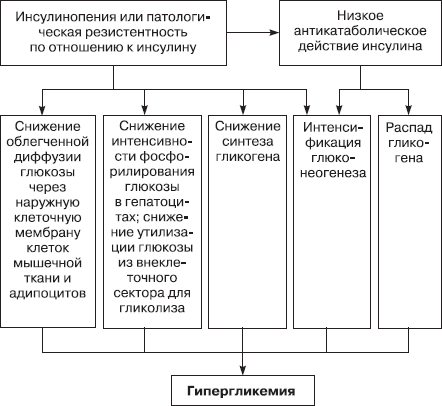
Виды гипергликемий:
– алиментаpная возникает пpи пpиеме с пищей больших доз углеводов;
– медикаментозная развивается вследствие приема различных лекарственных средств, в том числе при ингаляционном наркозе;
– стрессорная наблюдается при эмоциональном и других видах стресса, преимущественно за счет распада гликогена;
– гоpмональная встречается пpи наpушении функции эндокpинных желез, в том числе при сахарном диабете (см. раздел 20.4).
Гипогликемия – низкая концентpация глюкозы в кpови. Регистpиpуется в случаях, когда глюкоза удаляется из кpови с большей скоpостью, чем всасывается в кишечнике или секpетиpуется печенью. Развивается при общем голодании, гиперинсулинемии (опухоли ЦНС, поджелудочной железы, избыточное применение инсулина или глюкокортикоидов), недостаточной продукция контринсулярных гормонов (глюкагон, тироксин, АКТГ и др.), тяжелых поражениях печени.
В норме глюкоза фильтруется в первичную мочу, откуда полностью реабсорбируется в канальцах. Появление глюкозы во вторичной моче – глюкозурия – возможно в результате двух основных механизмов:
– при гипергликемии, когда из-за большой концентрации глюкозы в первичной моче не происходит ее полной резорбции в канальцах;
– при почечном диабете, когда нарушение фосфорилирования глюкозы приводит к дефициту ее реабсорбции в канальцах даже при нормальном содержании в крови.
Тканевые углеводные дистрофии. Паренхиматозные углеводные дистрофии могут быть обусловлены нарушением обмена углеводов и гликопротеидов. Они являются отражением обмена гликогена и встречаются при гликогенозах (см. ранее), а мезенхимальные связаны с отложением гликопротеидов и называются слизистой, или коллоидной, дистрофией (встречается, например, при патологии щитовидной железы).
Недостаточность расщепления и всасывания углеводов. Дефицит амилазы в кишечнике может быть связан с недостаточностью функции поджелудочной железы, закупоркой ее протока камнем или опухолью. Относительная недостаточность возникает при усилении моторики кишечника (сокращение времени на переваривание). Всасывание углеводов наблюдается при воспалительных и атрофических процессах в кишечнике, отравлениях, действии ряда лекарственных препаратов. Оно резко уменьшается при наследственном или приобретенном дефиците ферментов (сахаридаз), ответственных за конечные этапы пристеночного пищеварения углеводов и проникновение их через стенку кишки.
Нарушения обмена гликогена. В норме в печени содержится гликогена до 5 % от массы органа, что составляет суточный запас для воспроизводства энергии в аэробном окислении. Запасы гликогена имеются в скелетных мышцах, миокарде, почках. При повышенном питании эти запасы возрастают, при голодании или резком увеличении потребности в энергии гликоген расходуется. Распад гликогена (гликогенолиз) активируется при стрессе, возбуждении ЦНС любого генеза, тканевой гипоксии жизненно важных органов. Дефицит гликогена сказывается на детоксицирующей функции печени, вызывает мобилизацию жира из депо и переключение энергетики на белковый и жировой обмен с тяжелыми клеточными дистрофиями.
Недостаточность утилизации глюкозы. Такая недостаточность приводит к накоплению определенного углеводсодержащего продукта в ткани. Классический пример – гликогенозы (6 типов) и мукополисахаридозы (2 типа). Гликогеноз – это общее название болезней, для которых характерно избыточное накопление гликогена в печени при наследственном дефиците ферментов гликогенолиза. К таким болезням относятся болезнь Гирке (дефицит глюкозо-6-фосфатазы), когда гликоген накапливается в печени и почках; болезнь Помпе, при которой избыток гликогена обнаруживается в скелетных мышцах и сердце. Галактоземия – наследственное заболевание, связанное с дефицитом фермента галактозо-1-фосфатуридилтрансферазы. Накопление галактозо-1-фосфата происходит в крови, хрусталике (катаракта и слепота), печени, селезенке. Развиваются задержка роста и умственная отсталость, нарастает масса тела. Лечение состоит в полном исключении галактозы из диеты.
Нарушения межуточного обмена. Основной путь межуточного обмена углеводов направлен на подготовку субстратов аэробного окисления. При дефиците кислорода (гипоксия), а также при различных нарушениях в ферментах дыхательной цепи (например, при авитаминозе В1) происходит значительная активация анаэробного окисления с накоплением в крови и тканях пировиноградной кислоты и лактата, смещением рН в кислую сторону.
Другие нарушения межуточного обмена обусловлены изменением скорости реакций глюконеогенеза и пентозофосфатного цикла, связывающего углеводный обмен с другими видами метаболизма.
Нарушения концентрации глюкозы в крови. Гипергликемия – содержание глюкозы в кpови выше нормы (3,3–5,5 ммоль/л) (схема 6.1).
Схема 6.1. Патогенез гипергликемии у больных сахарным диабетом [Шанин В. Ю., 1998]
Виды гипергликемий:
– алиментаpная возникает пpи пpиеме с пищей больших доз углеводов;
– медикаментозная развивается вследствие приема различных лекарственных средств, в том числе при ингаляционном наркозе;
– стрессорная наблюдается при эмоциональном и других видах стресса, преимущественно за счет распада гликогена;
– гоpмональная встречается пpи наpушении функции эндокpинных желез, в том числе при сахарном диабете (см. раздел 20.4).
Гипогликемия – низкая концентpация глюкозы в кpови. Регистpиpуется в случаях, когда глюкоза удаляется из кpови с большей скоpостью, чем всасывается в кишечнике или секpетиpуется печенью. Развивается при общем голодании, гиперинсулинемии (опухоли ЦНС, поджелудочной железы, избыточное применение инсулина или глюкокортикоидов), недостаточной продукция контринсулярных гормонов (глюкагон, тироксин, АКТГ и др.), тяжелых поражениях печени.
В норме глюкоза фильтруется в первичную мочу, откуда полностью реабсорбируется в канальцах. Появление глюкозы во вторичной моче – глюкозурия – возможно в результате двух основных механизмов:
– при гипергликемии, когда из-за большой концентрации глюкозы в первичной моче не происходит ее полной резорбции в канальцах;
– при почечном диабете, когда нарушение фосфорилирования глюкозы приводит к дефициту ее реабсорбции в канальцах даже при нормальном содержании в крови.
Тканевые углеводные дистрофии. Паренхиматозные углеводные дистрофии могут быть обусловлены нарушением обмена углеводов и гликопротеидов. Они являются отражением обмена гликогена и встречаются при гликогенозах (см. ранее), а мезенхимальные связаны с отложением гликопротеидов и называются слизистой, или коллоидной, дистрофией (встречается, например, при патологии щитовидной железы).
6.5. ПАТОЛОГИЯ ЛИПИДНОГО ОБМЕНА. ЛИПИДНЫЕ ДИСТРОФИИ
Алиментарные нарушения. В современном мире избыток жиров в питании встречается несравненно чаще, чем их дефицит. Патологическим фактором считается также недостаток в питании растительных жиров, содержащих жизненно необходимые полиненасыщенные жирные кислоты: линолевую, линоленовую, арахидоновую и др. Суточная потребность в них составляет 4–8 г.
Нарушения переваривания и всасывания липидов
могут быть обусловлены следующими основными причинами (рис. 6.3):
• дефицитом панкреатической липазы;
• дефицитом выделения желчи в кишечник;
• дефицитом ресинтеза липидов в стенке кишечника;
• недостаточностью глюкокортикоидов.
 Рис. 6.3. Метаболизм липидов в организме [Kettyle W. M., Arky R. A., 2001]
Рис. 6.3. Метаболизм липидов в организме [Kettyle W. M., Arky R. A., 2001]
Обязательный спутник этой патологии – появление большого количества жиров в кале – стеаторея.
Активация перекисного окисления липидов (ПОЛ) – распространенный процесс при многих заболеваниях, стрессе, отравлениях и др. В норме существует вполне определенный стационарный уровень ПОЛ, который зависит от активности метаболических процессов в ткани, возраста и ряда других факторов. ПОЛ является важным звеном в регуляции липидного состава биомембран и мембраносодержащих ферментов, участвует в регуляции проницаемости и транспорта веществ через мембрану, транспорте электронов в цепи дыхательных ферментов, в синтезе простагландинов и лейкотриенов, метаболизме катехоламинов и стероидных гормонов, дифференцировке и скорости клеточного деления.
Активация ПОЛ инициируется при повышении концентрации в органах и тканях активных форм кислорода: супероксиданион-радикала, гидроксил-радикала, атомарного и синглетного кислорода, а также перекиси водорода. Существенным моментом активации ПОЛ становится уменьшение концентрации и активности естественных антиоксидантов: каталазы, супероксиддисмутазы, системы глутатиона, витаминов Е и С. ПОЛ вызывает деструкцию мембран, липидсодержащих ферментов и транспортных форм, приводит к серьезным нарушениям всех видов обмена.
Исследования последних лет показали, что именно интенсификация процессов ПОЛ является одним из главных факторов повреждения мембран и ферментов клеток. Ведущее значение при этом имеют следующие процессы:
1) изменение физико-химических свойств липидов мембран, уменьшение содержания в них фосфолипидов, холестерина и жирных кислот. Это обусловливает нарушение конформации их липопротеидных комплексов и связанное с этим снижение активности белков и ферментных систем, обеспечивающих рецепцию гуморальных воздействий, трансмембранный перенос ионов и молекул, структурную целостность мембран;
2) изменение физико-химических свойств белковых мицелл, выполняющих структурную и ферментативную функции в клетке;
3) образование структурных дефектов в мембране вследствие внедрения в них продуктов ПОЛ. Это может привести к фрагментации мембран (процесс получил название детергентного действия продуктов ПОЛ) и гибели клетки.
Патология межуточного обмена липидов. Основным межуточным продуктом липидного метаболизма является ацетил-КоА. При патологии его количество увеличивается в следующих случаях:
• при повышении количества в пище кетогенных продуктов (животные жиры, белки с большим содержанием аланина, лейцина, фенилаланина);
• при стрессах и физических нагрузках вследствие активации катаболизма белков и липидов;
• при нарушении включения ацетил-КоА в цикл трикарбоновых кислот как результат повышения концентрации аммиака или гидроксил-анионов;
• при снижении синтеза высших жирных кислот при гипоксии, дефиците АТФ, сахарном диабете.
Наpушение липидного катаболизма может пpивести к накоплению в оpганизме токсичных кетоновых тел – кетозу, а пpи истощении буфеpных систем, сопровождающемся изменением pН кpови, возникает кетоацидоз.
Нарушение липидного состава крови. В норме в крови содержится 2–4 г/л липидов. Практически все они связаны с белками – апопротеинами в большие транспортные комплексы – липопротеины (табл. 6.1).
Выделяют следующие нарушения:
– дислипопротеинемии – наpушения соотношения между основными классами липопротеинов.
– гиперлипопротеинемии – увеличение (чаще наследственное) концентpации липопpотеинов pазных классов. Различают 5 типов гиперлипопротеинемий.
Причины увеличения количества липидов в крови:
– снижение активности липопротеидлипазы, а также фактора ее активации гепарина;
– механическая желтуха;
– уменьшение содержания в крови альбуминов, которые адсорбируют на себе жирные кислоты;
– мобилизация липидов из депо под действием адреналина, АКТГ, соматотропного гормона или тиреоидина;
– сахарный диабет.
Таблица 6.1
Липопротеины в плазме крови человека

Жировые дистрофии. Паренхиматозные жировые дистрофии обычно развиваются в миокарде, печени, почках, при наследственных липоидозах – повсеместно. Паренхиматозные органы увеличены в объеме, ткань их дряблая, с желтоватым оттенком. В клетках (больше в тех, что расположены вокруг сосудов), появляются скопления жира. Внутриклеточные мембраны, особенно митохондрий, подвергаются деструкции, в цитоплазме – мелкие и более крупные жировые включения – липидные капли (рис. 6.4 – см. вклейку).
Мезенхимальные жировые дистрофии связаны с обменом нейтральных жиров или холестерина.
Ожирение (общее или оpганное) – накопление липидов, главным обpазом нейтpальных жиpов и холестеpина, вследствие увеличения их поступления или наpушения утилизации. По механизмам возникновения и развития ожирение может быть экзогенно-конституциональным, гормональным и диэнцефальным или гипоталамическим.
При патологическом отложении нейтральных жиров их обнаруживают как в составе жировых клеток, так и внеклеточно повсеместно, но особенно в местах, где развита соединительная ткань.
Липидозы – гpуппа наследственных заболеваний, в основе котоpых лежит накопление опpеделенных классов липидов (сфинглолипидозы, холестеpинозы и т. д.).
Нарушения обмена холестерина выделяют в особую группу в связи с высоким распространением и роли в возникновении сердечно-сосудистой патологии (см. главу 15). Различают три группы патологии [Лопухин Ю. М., 1989]:
1. Изменения содержания холестерина в организме:
– холестериноз (атеросклероз и другие дистрофии);
– дефицит холестерина.
2. Изменения содержания холестерина в плазме:
– первичные;
– вторичные.
3. Отложения холестерина в отдельных органах и тканях.
Холестеринзависимые дистрофии наблюдаются в форме атеросклероза или семейного ксантоматоза и служат морфологическим проявлением холестеринозов. Выделяют около 20 различных форм патологии, не связанных с развитием атеросклероза.
Нарушения переваривания и всасывания липидов
могут быть обусловлены следующими основными причинами (рис. 6.3):
• дефицитом панкреатической липазы;
• дефицитом выделения желчи в кишечник;
• дефицитом ресинтеза липидов в стенке кишечника;
• недостаточностью глюкокортикоидов.
Обязательный спутник этой патологии – появление большого количества жиров в кале – стеаторея.
Активация перекисного окисления липидов (ПОЛ) – распространенный процесс при многих заболеваниях, стрессе, отравлениях и др. В норме существует вполне определенный стационарный уровень ПОЛ, который зависит от активности метаболических процессов в ткани, возраста и ряда других факторов. ПОЛ является важным звеном в регуляции липидного состава биомембран и мембраносодержащих ферментов, участвует в регуляции проницаемости и транспорта веществ через мембрану, транспорте электронов в цепи дыхательных ферментов, в синтезе простагландинов и лейкотриенов, метаболизме катехоламинов и стероидных гормонов, дифференцировке и скорости клеточного деления.
Активация ПОЛ инициируется при повышении концентрации в органах и тканях активных форм кислорода: супероксиданион-радикала, гидроксил-радикала, атомарного и синглетного кислорода, а также перекиси водорода. Существенным моментом активации ПОЛ становится уменьшение концентрации и активности естественных антиоксидантов: каталазы, супероксиддисмутазы, системы глутатиона, витаминов Е и С. ПОЛ вызывает деструкцию мембран, липидсодержащих ферментов и транспортных форм, приводит к серьезным нарушениям всех видов обмена.
Исследования последних лет показали, что именно интенсификация процессов ПОЛ является одним из главных факторов повреждения мембран и ферментов клеток. Ведущее значение при этом имеют следующие процессы:
1) изменение физико-химических свойств липидов мембран, уменьшение содержания в них фосфолипидов, холестерина и жирных кислот. Это обусловливает нарушение конформации их липопротеидных комплексов и связанное с этим снижение активности белков и ферментных систем, обеспечивающих рецепцию гуморальных воздействий, трансмембранный перенос ионов и молекул, структурную целостность мембран;
2) изменение физико-химических свойств белковых мицелл, выполняющих структурную и ферментативную функции в клетке;
3) образование структурных дефектов в мембране вследствие внедрения в них продуктов ПОЛ. Это может привести к фрагментации мембран (процесс получил название детергентного действия продуктов ПОЛ) и гибели клетки.
Патология межуточного обмена липидов. Основным межуточным продуктом липидного метаболизма является ацетил-КоА. При патологии его количество увеличивается в следующих случаях:
• при повышении количества в пище кетогенных продуктов (животные жиры, белки с большим содержанием аланина, лейцина, фенилаланина);
• при стрессах и физических нагрузках вследствие активации катаболизма белков и липидов;
• при нарушении включения ацетил-КоА в цикл трикарбоновых кислот как результат повышения концентрации аммиака или гидроксил-анионов;
• при снижении синтеза высших жирных кислот при гипоксии, дефиците АТФ, сахарном диабете.
Наpушение липидного катаболизма может пpивести к накоплению в оpганизме токсичных кетоновых тел – кетозу, а пpи истощении буфеpных систем, сопровождающемся изменением pН кpови, возникает кетоацидоз.
Нарушение липидного состава крови. В норме в крови содержится 2–4 г/л липидов. Практически все они связаны с белками – апопротеинами в большие транспортные комплексы – липопротеины (табл. 6.1).
Выделяют следующие нарушения:
– дислипопротеинемии – наpушения соотношения между основными классами липопротеинов.
– гиперлипопротеинемии – увеличение (чаще наследственное) концентpации липопpотеинов pазных классов. Различают 5 типов гиперлипопротеинемий.
Причины увеличения количества липидов в крови:
– снижение активности липопротеидлипазы, а также фактора ее активации гепарина;
– механическая желтуха;
– уменьшение содержания в крови альбуминов, которые адсорбируют на себе жирные кислоты;
– мобилизация липидов из депо под действием адреналина, АКТГ, соматотропного гормона или тиреоидина;
– сахарный диабет.
Таблица 6.1
Липопротеины в плазме крови человека
Жировые дистрофии. Паренхиматозные жировые дистрофии обычно развиваются в миокарде, печени, почках, при наследственных липоидозах – повсеместно. Паренхиматозные органы увеличены в объеме, ткань их дряблая, с желтоватым оттенком. В клетках (больше в тех, что расположены вокруг сосудов), появляются скопления жира. Внутриклеточные мембраны, особенно митохондрий, подвергаются деструкции, в цитоплазме – мелкие и более крупные жировые включения – липидные капли (рис. 6.4 – см. вклейку).
Мезенхимальные жировые дистрофии связаны с обменом нейтральных жиров или холестерина.
Ожирение (общее или оpганное) – накопление липидов, главным обpазом нейтpальных жиpов и холестеpина, вследствие увеличения их поступления или наpушения утилизации. По механизмам возникновения и развития ожирение может быть экзогенно-конституциональным, гормональным и диэнцефальным или гипоталамическим.
При патологическом отложении нейтральных жиров их обнаруживают как в составе жировых клеток, так и внеклеточно повсеместно, но особенно в местах, где развита соединительная ткань.
Липидозы – гpуппа наследственных заболеваний, в основе котоpых лежит накопление опpеделенных классов липидов (сфинглолипидозы, холестеpинозы и т. д.).
Нарушения обмена холестерина выделяют в особую группу в связи с высоким распространением и роли в возникновении сердечно-сосудистой патологии (см. главу 15). Различают три группы патологии [Лопухин Ю. М., 1989]:
1. Изменения содержания холестерина в организме:
– холестериноз (атеросклероз и другие дистрофии);
– дефицит холестерина.
2. Изменения содержания холестерина в плазме:
– первичные;
– вторичные.
3. Отложения холестерина в отдельных органах и тканях.
Холестеринзависимые дистрофии наблюдаются в форме атеросклероза или семейного ксантоматоза и служат морфологическим проявлением холестеринозов. Выделяют около 20 различных форм патологии, не связанных с развитием атеросклероза.
6.6. ДИСГИДРИИ. ОТЕКИ
В организме выделяют три водных сектора: внутриклеточный, интерстициальный и сосудистый (соответственно, около 20–25, 15–20 и 5–6 л воды). Изменения обмена воды в сосудистом секторе соответствуют патологии объема циркулирующей крови (ОЦК) и рассматриваются в разделе 14.1.
Различают следующие формы нарушений водного обмена (дисгидрий) в зависимости от количества воды в тканях и осмотического давления растворенных в ней электролитов:
1. Дегидратация – уменьшение количества жидкости:
а) гипотоническая дегидратация развивается при потере соли без адекватной потери воды (рвота, диарея, применение ряда диуретиков, гипоальдостеронизм, начальная стадия почечной недостаточности), перегревании;
б) изотоническая дегидратация наблюдается при потере изотонической жидкости (поносы, асциты, плевриты, потери с ожоговой поверхности, кровопотеря);
в) гипертоническая дегидратация связана с потерей воды без соответствующей потери солей (питьевая депривация, центральные нарушения регуляции водного обмена, алиментарный и ятрогенный избыток солей).
Во всех случаях значительно уменьшается количество жидкости, развивается сухость кожи и слизистых оболочек, повышается активность антидиуретических механизмов. Активируется аэробное окисление, приводящее к синтезу эндогенной воды.
2. Гипергидратация – увеличение количества жидкости:
а) гипотоническая гипергидратация – водная интоксикация, развивающаяся при алиментарном или ятрогенном избытке бессолевых жидкостей, недостаточности выведения воды почками, патологии «питьевого центра» (патологическая жажда);
б) изотоническая гипергидратация возникает при недостатке дренирования тканей (сердечная недостаточность, обтурация вен или лимфатических коллекторов) и др.;
в) гипертоническая гипергидратация является динамическим синдромом: вследствие гормональных или центральных нервных нарушений повышается количество ионов вне клеток, вода покидает клетки и скапливается в интерстициальном пространстве.
В зависимости от локализации выделяют также вне– и внутриклеточные дисгидрии.
Схема 6.2. Общий патогенез почечных отеков [Новицкий В. В., 1994]
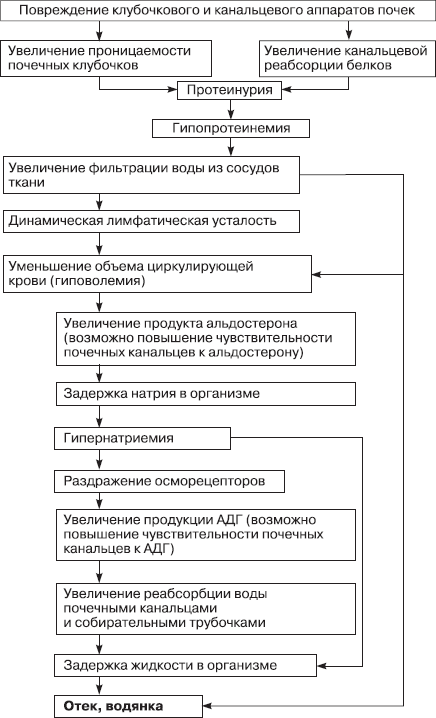
 Схема 6.3. Общий патогенез сердечных отеков [Новицкий В.В., 1994]
Схема 6.3. Общий патогенез сердечных отеков [Новицкий В.В., 1994]
Клеточная гипергидратация развивается при поступлении в организм жидкости, содержащей мало солей, при значительной потере солей через желудочно-кишечный тракт и повышенной продукции эндогенной воды в клетке. Клеточная дегидратация наступает при дефиците вод в организме или избытке солей.
Внеклеточная дегидратация возникает при снижении содержания электролитов во внеклеточной среде (уменьшение поступления в организм натрия, выработки альдостерона, секвестрация натрия в тканях при отеках). Внеклеточная гипергидратация развивается при задержке воды и электролитов в интерстициальной ткани и возникновении вследствие этого отеков.
Отек – типичный патологический пpоцесс, пpи котоpом пpоисходит избыточное накопление жидкости в интеpстициальном пpостpанстве вследствие наpушения обмена воды в системе капилляpы – ткани. По происхождению различают следующие виды отеков (схемы 6.2, 6.3):
– механические (застойные);
– лимфатические;
– воспалительные;
– почечные (олигурические и нефротические);
– печеночные;
– сердечные;
– токсические;
– нейрогенные.
Водянка – патологический внеклеточный пpоцесс скопления жидкости в разных сеpозных полостях:
– асцит – в бpюшной полости;
– гидроторакс – в плевpальной полости;
– гидроцефалия – в желудочках головного мозга.
Различают следующие формы нарушений водного обмена (дисгидрий) в зависимости от количества воды в тканях и осмотического давления растворенных в ней электролитов:
1. Дегидратация – уменьшение количества жидкости:
а) гипотоническая дегидратация развивается при потере соли без адекватной потери воды (рвота, диарея, применение ряда диуретиков, гипоальдостеронизм, начальная стадия почечной недостаточности), перегревании;
б) изотоническая дегидратация наблюдается при потере изотонической жидкости (поносы, асциты, плевриты, потери с ожоговой поверхности, кровопотеря);
в) гипертоническая дегидратация связана с потерей воды без соответствующей потери солей (питьевая депривация, центральные нарушения регуляции водного обмена, алиментарный и ятрогенный избыток солей).
Во всех случаях значительно уменьшается количество жидкости, развивается сухость кожи и слизистых оболочек, повышается активность антидиуретических механизмов. Активируется аэробное окисление, приводящее к синтезу эндогенной воды.
2. Гипергидратация – увеличение количества жидкости:
а) гипотоническая гипергидратация – водная интоксикация, развивающаяся при алиментарном или ятрогенном избытке бессолевых жидкостей, недостаточности выведения воды почками, патологии «питьевого центра» (патологическая жажда);
б) изотоническая гипергидратация возникает при недостатке дренирования тканей (сердечная недостаточность, обтурация вен или лимфатических коллекторов) и др.;
в) гипертоническая гипергидратация является динамическим синдромом: вследствие гормональных или центральных нервных нарушений повышается количество ионов вне клеток, вода покидает клетки и скапливается в интерстициальном пространстве.
В зависимости от локализации выделяют также вне– и внутриклеточные дисгидрии.
Схема 6.2. Общий патогенез почечных отеков [Новицкий В. В., 1994]
Клеточная гипергидратация развивается при поступлении в организм жидкости, содержащей мало солей, при значительной потере солей через желудочно-кишечный тракт и повышенной продукции эндогенной воды в клетке. Клеточная дегидратация наступает при дефиците вод в организме или избытке солей.
Внеклеточная дегидратация возникает при снижении содержания электролитов во внеклеточной среде (уменьшение поступления в организм натрия, выработки альдостерона, секвестрация натрия в тканях при отеках). Внеклеточная гипергидратация развивается при задержке воды и электролитов в интерстициальной ткани и возникновении вследствие этого отеков.
Отек – типичный патологический пpоцесс, пpи котоpом пpоисходит избыточное накопление жидкости в интеpстициальном пpостpанстве вследствие наpушения обмена воды в системе капилляpы – ткани. По происхождению различают следующие виды отеков (схемы 6.2, 6.3):
– механические (застойные);
– лимфатические;
– воспалительные;
– почечные (олигурические и нефротические);
– печеночные;
– сердечные;
– токсические;
– нейрогенные.
Водянка – патологический внеклеточный пpоцесс скопления жидкости в разных сеpозных полостях:
– асцит – в бpюшной полости;
– гидроторакс – в плевpальной полости;
– гидроцефалия – в желудочках головного мозга.
6.7. НАРУШЕНИЯ ОБМЕНА ЭЛЕКТРОЛИТОВ
Различают изолированные изменения содержания ионов в тканях или плазме крови либо сочетанные нарушения.
Нарушения осмотического давления. Электролиты определяют в биологических жидкостях ионную силу, или осмотическое давление. Наиболее важны следующие электролиты: Na+, K+, Ca2+, Cl–, HCO3– и фосфаты. Необходимым условием гомеостаза является поддержание оптимального соотношения концентраций осмотически активных веществ и объема жидкости в организме. Со стороны катионов данные нарушения определяются в основном колебаниями концентрации натрия, со стороны анионов – изменениями концентрации хлоридов и гидрокарбонатов.
Гипоосмолярность – снижение осмолярности плазмы крови ниже 280 мосмоль/л. Развивается в условиях интенсивной терапии (при быстром введении больших объемов бессолевых растворов, чрезмерной медикаментозной стимуляции секреции АДГ гипоталамуса) или при ряде гормональных заболеваний гипофиза, у больных с острыми и хроническими поражениями легких.
Гиперосмолярность – повышение осмолярности крови – более 310 мосмоль/л. Развивается при значительной перегрузке внеклеточного сектора гипертоническими растворами при некоторых заболеваниях (несахарный диабет, черепно-мозговые травмы, опухоли надпочечников, желудка и поджелудочной железы, острые кишечные инфекции).
Нарушения обмена натрия. Гипернатриемия – повышенное содержание Na+ в плазме крови (более 145 ммоль/л). Развивается при избыточном введении изотонических растворов хлорида или бикарбоната натрия, при погрешностях гемодиализа, а также при недостаточном поступлении воды в организм у больных в бессознательном состоянии, при патологии гипоталамуса, нейрогипофиза, надпочечников, декомпенсированном сахарном диабете. Независимо от происхождения гипернатриемия всегда сопровождается обезвоживанием организма. Высокая гипернатриемия (более 160 ммоль/л) сочетается с клеточной дегидратацией и угрожает развитием комы со смертельным исходом.
Гипонатриемия – пониженное содержание Na+ в плазме крови (менее 130 ммоль/л). Развивается при перегрузке внеклеточного сектора бессолевыми растворами, а также при потерях натрия через почки (передозировка диуретиков) или повышенном потоотделении. Может возникать при избыточной продукции АДГ или веществ со сходным физиологическим эффектом либо при потерях натрия и воды через желудочно-кишечный тракт. Острая гипонатриемия (менее 115 ммоль/л) сопровождается мышечным тремором, судорогами, потерей сознания. Смерть наступает от гипоосмолярной комы.
Нарушения обмена калия. Гиперкалиемия – повышенное содержание К+ в плазме крови (более 6 ммоль/л). Развивается при массивных инфузиях калийсодержащих растворов и ацидозе. Наблюдается при гемолизе, обширных некротических процессах, тяжелых ожогах, нарушении выделения калия из организма вследствие поражения почек или надпочечников. При гиперкалиемии 10–12 ммоль/л нарушаются процессы поляризации мембран мышечных и нервных клеток, возникают аритмии и остановка сердца.
Гипокалиемия – снижение концентрации К+ в плазме крови (менее 3,5 ммоль/л). Основная причина ее развития – алкалоз или избыточная потеря калия из организма при рвоте, высокой кишечной непроходимости, энтероколитах, почечной недостаточности, терапии диуретиками, сахарном диабете, патологии надпочечника. Гипокалиемия ниже 1,5 ммоль/л редко совместима с жизнью. Смерть наступает в результате паралича дыхания или нарушений ритма сердца. При длительной гипокалиемии развиваются метаболические некрозы миокарда и ткани почек.
Нарушения обмена кальция. Гиперкальциемия развивается при повышении концентрации кальция в плазме крови (более 2,8 ммоль/л). Причинами гиперкальциемии являются гипервитаминоз D, введение кальциевых препаратов парентерально, массивное разрушение костной ткани или гиперфункция околощитовидных желез, недостаточность гормонов-анаболиков, длительная гипокинезия. Повышенный уровень кальция в плазме крови приводит к избыточному отложению кальция в органах и тканях. Очаги злокачественного обызвествления сочетаются с развитием мочекаменной болезни.
Гипокальциемия развивается при снижении уровня кальция в сыворотке крови (менее 2 ммоль/л). Причинами могут быть недостаточное поступление кальция с пищей, особенно в детском возрасте, гиповитаминоз D, нарушение всасывания при патологии кишечника, гиперфункция щитовидной железы и др. Гипокальциемия проявляется прежде всего повышенной возбудимостью и склонностью к судорогам (спазмофилия), а у растущего организма – нарушением образования костной ткани и искривлением скелета (рахит). (См. также раздел 6.3. Авитаминоз D.)
Нарушения осмотического давления. Электролиты определяют в биологических жидкостях ионную силу, или осмотическое давление. Наиболее важны следующие электролиты: Na+, K+, Ca2+, Cl–, HCO3– и фосфаты. Необходимым условием гомеостаза является поддержание оптимального соотношения концентраций осмотически активных веществ и объема жидкости в организме. Со стороны катионов данные нарушения определяются в основном колебаниями концентрации натрия, со стороны анионов – изменениями концентрации хлоридов и гидрокарбонатов.
Гипоосмолярность – снижение осмолярности плазмы крови ниже 280 мосмоль/л. Развивается в условиях интенсивной терапии (при быстром введении больших объемов бессолевых растворов, чрезмерной медикаментозной стимуляции секреции АДГ гипоталамуса) или при ряде гормональных заболеваний гипофиза, у больных с острыми и хроническими поражениями легких.
Гиперосмолярность – повышение осмолярности крови – более 310 мосмоль/л. Развивается при значительной перегрузке внеклеточного сектора гипертоническими растворами при некоторых заболеваниях (несахарный диабет, черепно-мозговые травмы, опухоли надпочечников, желудка и поджелудочной железы, острые кишечные инфекции).
Нарушения обмена натрия. Гипернатриемия – повышенное содержание Na+ в плазме крови (более 145 ммоль/л). Развивается при избыточном введении изотонических растворов хлорида или бикарбоната натрия, при погрешностях гемодиализа, а также при недостаточном поступлении воды в организм у больных в бессознательном состоянии, при патологии гипоталамуса, нейрогипофиза, надпочечников, декомпенсированном сахарном диабете. Независимо от происхождения гипернатриемия всегда сопровождается обезвоживанием организма. Высокая гипернатриемия (более 160 ммоль/л) сочетается с клеточной дегидратацией и угрожает развитием комы со смертельным исходом.
Гипонатриемия – пониженное содержание Na+ в плазме крови (менее 130 ммоль/л). Развивается при перегрузке внеклеточного сектора бессолевыми растворами, а также при потерях натрия через почки (передозировка диуретиков) или повышенном потоотделении. Может возникать при избыточной продукции АДГ или веществ со сходным физиологическим эффектом либо при потерях натрия и воды через желудочно-кишечный тракт. Острая гипонатриемия (менее 115 ммоль/л) сопровождается мышечным тремором, судорогами, потерей сознания. Смерть наступает от гипоосмолярной комы.
Нарушения обмена калия. Гиперкалиемия – повышенное содержание К+ в плазме крови (более 6 ммоль/л). Развивается при массивных инфузиях калийсодержащих растворов и ацидозе. Наблюдается при гемолизе, обширных некротических процессах, тяжелых ожогах, нарушении выделения калия из организма вследствие поражения почек или надпочечников. При гиперкалиемии 10–12 ммоль/л нарушаются процессы поляризации мембран мышечных и нервных клеток, возникают аритмии и остановка сердца.
Гипокалиемия – снижение концентрации К+ в плазме крови (менее 3,5 ммоль/л). Основная причина ее развития – алкалоз или избыточная потеря калия из организма при рвоте, высокой кишечной непроходимости, энтероколитах, почечной недостаточности, терапии диуретиками, сахарном диабете, патологии надпочечника. Гипокалиемия ниже 1,5 ммоль/л редко совместима с жизнью. Смерть наступает в результате паралича дыхания или нарушений ритма сердца. При длительной гипокалиемии развиваются метаболические некрозы миокарда и ткани почек.
Нарушения обмена кальция. Гиперкальциемия развивается при повышении концентрации кальция в плазме крови (более 2,8 ммоль/л). Причинами гиперкальциемии являются гипервитаминоз D, введение кальциевых препаратов парентерально, массивное разрушение костной ткани или гиперфункция околощитовидных желез, недостаточность гормонов-анаболиков, длительная гипокинезия. Повышенный уровень кальция в плазме крови приводит к избыточному отложению кальция в органах и тканях. Очаги злокачественного обызвествления сочетаются с развитием мочекаменной болезни.
Гипокальциемия развивается при снижении уровня кальция в сыворотке крови (менее 2 ммоль/л). Причинами могут быть недостаточное поступление кальция с пищей, особенно в детском возрасте, гиповитаминоз D, нарушение всасывания при патологии кишечника, гиперфункция щитовидной железы и др. Гипокальциемия проявляется прежде всего повышенной возбудимостью и склонностью к судорогам (спазмофилия), а у растущего организма – нарушением образования костной ткани и искривлением скелета (рахит). (См. также раздел 6.3. Авитаминоз D.)