Страница:
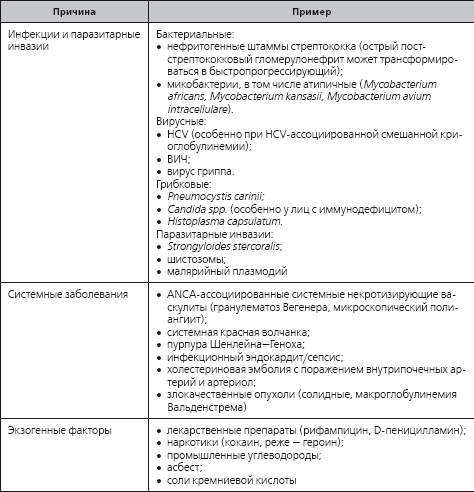
Известно также, что в одних и тех же условиях (например, при холестериновой эмболии внутрипочечных артериол) почечный процесс может развиваться как типичным образом (острый «аллергический» эозинофильный тубуло-интерстициальный нефрит с острой почечной недостаточностью), так и с ускоренным формированием быстропрогрессирующего гломерулонефрита, в том числе ANCA-ассоциированного. В связи с этим нельзя полностью исключить, что к быстропрогрессирующему гломерулонефриту существует определенная наследственная предрасположенность, однако уже в настоящее время очевидно, что конкретные генетические детерминанты трудно установить, и само по себе их носительство не является достаточным для развития почечного поражения. Тем не менее у больных антительным (тип I) анти-БМК-ассоциированным быстропрогрессирующим гломерулонефритом в рамках синдрома Гудпасчера удавалось констатировать значительную частоту обнаружения некоторых классов антигенов гистосовместимости (HLA DRB1-15, HLA DQB1-6, HLA-DRW 2). Можно тем не менее утверждать, что фенотипическому проявлению генетических детерминант быстропрогрессирующего гломерулонефрита, в том числе развивающегося в рамках синдрома Гудпасчера, почти всегда способствует действие экзогенных факторов, устранение которых зачастую не сопряжено с увеличением вероятности достижения ремиссии почечного поражения.
Таблица 3.1
Этиология быстропрогрессирующего гломерулонефрита
 Патогенез
Патогенез
Особенности клинической эволюции и прогноза быстропрогрессирующего гломерулонефрита во многом определяются типом участвующих в почечном поражении антител и вариантом их взаимодействия с гломерулярной базальной мембраной. Именно поэтому общепризнано разделение быстропрогрессирующего гломерулонефрита на иммунопатогенетические варианты (табл. 3.2), определяемые на основании оцениваемого при иммунофлуоресцентной микроскопии типа свечения антител в образцах ткани почки, полученной при биопсии, и наличия определенных сывороточных маркеров (антител к цитоплазме нейтрофилов (ANCA), анти-БМК-антител).
Таблица 3.2
Иммунопатогенетические типы быстропрогрессирующего нефрита
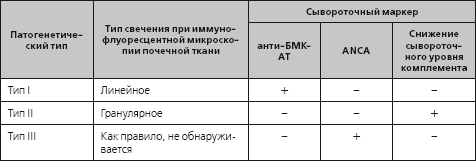 Тип I (антительный, анти-БМК-нефрит) cуществует изолированно или сочетается с поражением легких (легочно-почечный синдром, синдром Гудпасчера). Циркулирующие антитела к базальной мембране клубочка доступны для определения в сыворотке крови.
Тип I (антительный, анти-БМК-нефрит) cуществует изолированно или сочетается с поражением легких (легочно-почечный синдром, синдром Гудпасчера). Циркулирующие антитела к базальной мембране клубочка доступны для определения в сыворотке крови.
При II (иммунокомплексном) типе депозиты иммунных комплексов обнаруживают в мезангии и стенке капилляров почечного клубочка. Характерные серологические маркеры отсутствуют, за исключением сниженного сывороточного уровня комплемента, который может косвенно указывать на связь быстропрогрессирующего гломерулонефрита с HCV-ассоциированной смешанной криоглобулинемией или системной красной волчанкой.
Почечное поражение при III (малоиммунном) типе быстропрогрессирующего гломерулонефрита определяется клеточными иммунными реакциями, индуцируемыми ANCA. Быстропрогрессирующий гломерулонефрит III типа может быть локально-почечным, или, сочетаясь с поражением других органов, существовать как одно из проявлений системных ANCA-ассоциированных некротизирующих васкулитов (гранулематоз Вегенера, микроскопический полиангиит).
Более половины всех случаев быстропрогрессирующего гломерулонефрита относится к III типу, частота встречаемости I и II типов примерно одинакова (20–25 %).
Одним из центральных звеньев патогенеза быстропрогрессирующего экстракапиллярного гломерулонефрита с полулуниями считают проникновение белков плазмы крови и воспалительных клеток в пространство капсулы Шумлянского—Боумена. Накопление макрофагов вследствие миграции их предшественников из циркулирующей крови и пролиферации резидентных клеток дополняется миграцией из почечного интерстиция фибробластов и миофибробластов. Эти клетки начинают в избытке продуцировать компоненты экстрацеллюлярного матрикса (фибронектин, коллагены I и III типов), что способствует значительному повышению интенсивности фиброза полулуний, лежащего в основе необратимого ухудшения фильтрационной функции почек. Следует тем не менее подчеркнуть, что далеко не всегда при быстропрогрессирующем гломерулонефрите удается установить четкое соответствие между выраженностью почечной недостаточности и количеством полулуний, подвергшихся фибротической трансформации. С одной стороны, суммарное число почечных клубочков, в которых выявляются полулуния, можно считать решающим в развитии быстропрогрессирующего гломерулонефрита. Однако его формирование регистрируют и тогда, когда число клубочковых полулуний не достигает 50 % или они практически отсутствуют.
Межклеточным взаимодействиям в полулуниях, регулируемым определенными хемокинами (моноцитарным хемотаксическим протеином I типа (MCP-1), макрофагальным воспалительным протеином типа 1a (MIP1a)), очевидно, принадлежит решающая роль в формировании быстропрогрессирующего гломерулонефрита. Так, при экстракапиллярном гломерулонефрите установлена прямая корреляция между выраженностью фиброза полулуний и количеством экспрессирующих a-гладкомышечный актин миофибробластов в интерстиции в местах, близких к точкам разрыва капсулы Боумена; там же, а также в интерстициальных клетках, окружающих сосуды, с наибольшей интенсивностью синтезируется коллаген III типа. Трансформирующий фактор роста b-1 при экстракапиллярном гломерулонефрите обнаруживают преимущественно в канальцевых эпителиоцитах. Число миофибробластов, экспрессирующих a-гладкомышечный актин, и количество коллагена IV в почечном интерстиции позволяют предсказать вероятность необратимого ухудшения функции почек и ответ на иммуносупрессивную терапию. Показано, что нарастание экспрессии миофибробластов ассоциировано с усилением процессов апоптоза клеток почечного тубуло-интерстиция с одновременным увеличением синтеза медиаторов фиброгенеза, в первую очередь трансформирующего фактора роста-b.
Установлено, что при экстракапиллярном гломерулонефрите интенсивная экспрессия трансформирующего фактора роста-b ассоциирована с увеличением числа полулуний, подвергшихся фибротической трансформации. Более того, мочевая экскреция трансформирующего фактора роста-b у больных экстракапиллярным гломерулонефритом, не ответивших на иммуносупрессивную терапию и продемонстрировавших быстрое развитие терминальной почечной недостаточности, не менее чем в 2,5 раза превосходит таковую у тех пациентов, у кого прогрессирование заболевания удалось задержать с помощью иммуносупрессивной терапии.
Роль избытка трансформирующего фактора роста-b в прогрессировании быстропрогрессирующего экстракапиллярного гломерулонефрита, очевидно, не исчерпывается только его участием в процессах почечного фиброгенеза. Хорошо известно, что трансформирующий фактор роста-b – один из наиболее мощных физиологических индукторов клеточного апоптоза. Высокая интенсивность синтеза этого медиатора при экстракапиллярном гломерулонефрите может отражать блокаду соответствующих рецепторов и связанное с ней торможение процессов апоптоза, представляющее собой один из механизмов формирования полулуний. В норме трансформирующий фактор роста-b индуцирует клеточный апоптоз путем взаимодействия со специфическим рецептором BMRP (bone morphogenetic protein receptor – рецептором костного морфогенетического белка). Установлено, что при малоиммунном (тип III) варианте быстропрогрессирующего гломерулонефрита в почечных клубочках и интерстиции, в том числе в области полулуний, существенно возрастает экспрессия белка гремлина – антагониста BMRP, препятствующего таким образом взаимодействию этого белкового рецептора с трансформирующим фактором роста-b. В результате блокады BMRP гремлином миофибробласты и фибробласты не вступают в процесс апоптоза, а продолжают пролиферировать и дифференцироваться, что приводит к дальнейшему образованию новых полулуний и их интенсивному фиброзу, несмотря на возникающую по механизму отрицательной обратной связи гиперпродукцию трансформирующего фактора роста-b, который в этой ситуации начинает реализовывать свое профиброгенное действие другими путями, например через провокацию экспрессии медиаторов эндотелий-зависимого звена гемостаза (ингибитора активатора плазминогена-1).
Представление о быстропрогрессирующем гломерулонефрите как о заболевании, в основе развития которого лежит патология индуцируемого трансформирующим фактором роста-b апоптоза, открывает новые перспективы в лечении этого заболевания. Наряду с ингибированием блокирующего рецепторы BMRP гремлина можно рассчитывать на терапевтическую эффективность введения в почечную ткань доноров BMRP, в том числе клеток костного мозга. Показано, что костный мозг может быть источником почечных мезангиоцитов и эндотелиоцитов, трансдифференциация которых осуществляется в условиях характерного микроокружения и при участии некоторых факторов роста, в частности тромбоцитарного. Гиперпродукция блокаторов BMRP или недостаточная экспрессия этих рецепторов могут рассматриваться как одно из наиболее важных обстоятельств, предрасполагающих к развитию именно быстропрогрессирующего экстракапиллярного гломерулонефрита. Более того, сама по себе недостаточность рецепторов BMRP может быть генетически детерминированной (аналогично первичной легочной гипертензии – другому заболеванию, в основе которого лежит патология апоптоза отдельных клеточных пулов с их избыточной пролиферацией, при котором носительство мутантных форм гена BMRP четко продемонстрировано и обусловливает семейные случаи). Следовательно, изучение вариантов гена BMRP и интенсивности экспрессии его блокаторов (в частности, гремлина) может способствовать более детальному пониманию процессов, лежащих в основе развития быстропрогрессирующего гломерулонефрита, в том числе объяснению его развития у лиц, исходно страдающих другим хроническим заболеванием почек, например диабетической нефропатией.
Понятно, что каждый из типов быстропрогрессирующего гломерулонефрита имеет определенные патогенетические особенности.
Развитие синдрома Гудпасчера (тип I – антительный – быстропрогрессирующего гломерулонефрита) связывают со специфическими анти-БМК антителами, связывающимися с базальной мембраной альвеол и почечного клубочка и обладающими высокой специфичностью к a-3 цепи коллагена IV типа. Cвязь анти-БМК антител с a-3 цепью коллагена IV типа происходит через содержащийся в ней так называемый неколлагеновый (NC) домен. Cуществует 4 типа анти-БМК антител (А, В, АВ, Х). А и В типы взаимодействуют с определенными эпитопами (А или В, соответственно), АВ – с обоими названными эпитопами, точка приложения действия анти-БМК антител Х типа неясна. Все эти эпитопы расположены на NC домене.
В настоящее время синдром Гудпасчера продолжают интенсивно изучать на экспериментальных моделях: создан синтетический аналог соответствующего антигена, при введении которого животным у них развивается легочно-почечный синдром, обязательно включающий антительный быстропрогрессирующий гломерулонефрит. У крыс с синдромом Гудпасчера удалось детально проследить эволюцию почечного поражения. Депозиты IgG на базальной мембране формируются через 2 недели после иммунизации, одновременно констатируют отек клубочковых эндотелиоцитов и появление альбуминурии. Клеточный инфильтрат представлен преимущественно СD4-позитивными Т-лимфоцитами. На 3-й неделе начинают образовываться полулуния, в капсуле Боумена накапливается большое количество фибрина, гломерулярный инфильтрат представлен преимущественно CD8-позитивными Т-лимфоцитами и макрофагами. Характерны множественные разрывы базальной мембраны.
Показано, что в формировании тканевого повреждения при синдроме Гудпасчера существенную роль играют Т-лимфоциты. Продемонстрировано значительное нарастание числа CD4-позитивных Т-лимфоцитов, распознающих NC-домен a-3 цепи коллагена IV типа в периферической крови в острую фазу заболевания. Если обострение удается прервать, число их существенно уменьшается. Развитие синдрома Гудпасчера может быть также обусловлено нарушениями регуляции экспрессии IgG. Так, в эксперименте удавалось воспроизвести синдром Гудпасчера у мышей с отсутствующим Fc-гамма рецептором IIB, играющим центральную роль в контроле экспрессии IgG. Часть случаев синдрома Гудпасчера может также быть обусловлена моноклональной пролиферацией IgA.
При синдроме Гудпасчера фибробласты почечного тубуло-интерстиция активно экспрессируют a-гладкомышечный актин, в то время как виментина и трансформирующего фактора роста-b на их поверхности обнаружить не удается. Показано, что блокада моноцитарного хемоаттрактантного протеина I типа позволяет уменьшить выраженность тубуло-интерстициального повреждения при синдроме Гудпасчера, что также открывает определенные перспективы в лечении данного заболевания.
Иммунокомплексный (тип II) вариант быстропрогрессирующего гломерулонефрита развивается при наличии соответствующего субстрата для образования иммунных комплексов. В качестве такового могут выступать бактериальные антигены, лекарственные препараты или их метаболиты, криоглобулины при HCV-ассоциированной смешанной криоглобулинемии, антитела к ДНК при системной красной волчанке.
Почечное поражение при III (малоиммунном, pauci) типе быстропрогрессирующего гломерулонефрита определяется тканьдеструктивным действием ANCA, степень повышения сывороточной концентрации которых, как правило, отражает активность заболевания. Взаимодействие между Fc-рецепторами нейтрофилов, миелопероксидазой, протеинкиназой-3 и ANCA сопровождается гиперпродукцией супероксид-иона, перекисей, а также провоспалительных цитокинов. Одновременно возрастает экспрессия b2-интегрина – молекулы адгезии, принимающей непосредственное участие во взаимодействии нейтрофилов с эндотелиоцитами (повреждение эндотелиоцитов при ANCA-ассоциированном быстропрогрессирующем гломерулонефрите может также быть связано с продукцией антиэндотелиальных антител). В тканях-мишенях ANCA формируются также олигоклональные пулы Т-лимфоцитов. Эти Т-лимфоциты, модулируя продукцию интерферона-g и a-фактора некроза опухолей, принимают непосредственное участие в образовании полулуний. Более тяжелое почечное поражение наблюдают при преобладании CD28– Т-лимфоцитов.
Детали патогенеза отдельных вариантов быстропрогрессирующего гломерулонефрита продолжают уточняться. Для разработки селективных терапевтических воздействий необходимо уточнение механизмов формирования полулуний и индукторов их фибротической трансформации.
Морфология
Гломерулонефрит с полулуниями (быстропрогрессирующий, подострый, злокачественный, экстракапиллярный пролиферативный) – тяжелая форма гломерулонефрита, при которой в большинстве клубочков формируются эпителиальные полулуния.
Важный патогенетический фактор формирования полулуний в пространстве Боумена – выход фибрина в просвет капсулы почечного тельца. Вероятно, фибрин и другие белки плазмы крови, а также моноциты попадают в просвет капсулы почечного тельца через разрывы в воспаленных капиллярах клубочков. Фибрин и его компоненты являются стимулом для миграции моноцитов. Макрофаги, образующиеся из моноцитов, пролиферирующие париетальные эпителиальные клетки и полиморфно-ядерные лейкоциты образуют «клеточные» полулуния. Эти полулуния постепенно сдавливают капиллярный клубочек, серьезно затрудняя и даже прекращая образование первичной мочи с развитием олигоурии и анурии. Спавшиеся капиллярные петли пропитываются фибрином. По мере прогрессирования процесса полулуния приобретают фиброзный характер, в них появляются фибробласты и коллаген. В финале клубочек полностью склерозируется.
Гломерулонефрит с полулуниями в большой степени является результатом клеточно-опосредованного иммунного повреждения клубочков. В развитии данного заболевания определенная роль отводится гиперчувствительности замедленного типа (ГЗТ), при которой сенсибилизированные Т-лимфоциты привлекают и активируют макрофаги. При этом отмечается значительная локальная экспрессия тканевого фактора в почечных клубочках. Наблюдается выраженная гломерулярная аккумуляция CD4+ Т-лимфоцитов. Обнаружение CD8+ Т-лимфоцитов свидетельствует об участии Т-клеточно-опосредованной цитотоксичности в патогенезе гломерулонефрита с полулуниями. Цитотоксические Т-лимфоциты вызывают повреждение как секретируя цитотоксические молекулы, так и с помощью клеточных медиаторов (фактора некроза опухолей a-ФНО и Fas-лиганды), взаимодействующих со своими рецепторами на клетках-мишенях и вызывающих их апоптоз. Известно, что в интрагломерулярных мононуклеарных клетках происходит усиленная продукция РНК провоспалительных цитокинов ФНО-a и интерлейкина-1b (ИЛ-1b). Считается, что ИЛ-1b продуцируют в основном макрофаги в полулуниях, в то время как ФНОa продуцируется клетками почечных клубочков, в том числе мезангиальных, и эпителиальными клетками почечных канальцев. Продукция ИЛ-1b и его взаимодействие со своим рецептором I типа на клетках почечных клубочков вызывает экспрессию ФНО-a и дальнейшее повреждение клубочков.
Важную роль в регуляции макрофагальной инфильтрации почечных клубочков при гломерулонефрите с полулуниями играет моноцитарный хемоаттрактантный белок-1 (MCP-1) и его рецепторы. В почечных клубочках отмечается усиленная экспрессия MCP-1 клетками полулуний, париетальными эпителиальными клетками, эпителием канальцев и инфильтрирующими интерстиций лейкоцитами. Также возрастает экспрессия хемокинового рецептора 2В, обнаруживаемого в основном на мононуклеарных лейкоцитах, инфильтрирующих клубочек и полулуния. В привлечении лейкоцитов и формировании инфильтрата определенную роль играют адгезивные молекулы ICAM-1 и VCAM-1, повышенная экспрессия которых выявляется в пораженных почечных клубочках пациентов с гломерулонефритом с полулуниями (анти-ГБМ и малоиммунном типах).
Макроскопически при быстропрогрессирующем гломерулонефрите почки увеличены в размерах, дряблые; кора широкая, желто-серая, тусклая с красным крапом, резко отграничена от темно-красных пирамид – «большая пестрая почка», или красная, сливается с пирамидами – «большая красная почка».
В гистологической картине доминирует образование полулуний, которые могут занимать только отдельные сегменты пространства капсулы почечного тельца или полностью окружать клубочек. Развитие полулуний проходит несколько стадий: от клеточных полулуний к фиброзно-клеточным, а затем – фиброзным. Как уже было сказано, «клеточные» полулуния, занимающие пространство между капсулой и капиллярным клубочком, представлены пролиферирующими париетальными эпителиальными клетками и макрофагами, также в них обнаруживаются нейтрофилы и лимфоциты. В течение нескольких недель в полулуниях развиваются процессы организации с формированием соединительнотканного матрикса, который хорошо выявляется при серебрении. Полулуния, полностью окружающие клубочки, вызывают тотальный склероз. Наблюдаются также отек, инфильтрация и склероз стромы, атрофия канальцев.
Быстропрогрессирующий гломерулонефрит подразделяют на 3 группы: анти-ГБМ гломерулонефрит; иммунокомплексный гломерулонефрит с полулуниями; малоиммунный гломерулонефрит с полулуниями.
Анти-ГБМ гломерулонефрит характеризуется линейными отложениями IgG и С3 компонента комплемента вдоль гломерулярной базальной мембраны. Вызывается аутоантителами к a-3 цепи коллагена IV типа. Составляет от 10 до 20 % случаев гломерулонефрита с полулуниями. Анти-ГБМ гломерулонефрит может ограничиваться поражением почек или развиваться в форме почечно-легочного синдрома. Классическим примером последнего является быстропрогрессирующий гломерулонефрит, связанный с синдромом Гудпасчера. Антитела к БМК перекрестно реагируют с базальными мембранами легочных альвеол. Обусловливают появление клинической картины легочных кровоизлияний, сопровождающихся почечной недостаточностью. Кроме того, анти-ГБМ гломерулонефрит может быть осложнением других заболеваний, например развиваться de novo после трансплантации почки у пациентов с синдромом Альпорта, а также обнаруживаться у пациентов с мембранозным гломерулонефритом с образованием полулуний. Примерно у 1/3 пациентов с анти-ГБМ гломерулонефритом выявляются циркулирующие антинейтрофильные цитоплазматические антитела (ANCA), и данное заболевание у них ассоциировано с васкулитом мелких сосудов различных органов (не только почек и легких).
Типичной картиной при световой микроскопии является некротизирующий гломерулонефрит с очаговым или диффузным формированием полулуний. В пораженных участках капилляров почечных клубочков обнаруживаются скопления лейкоцитов. Непораженные участки могут иметь нормальный вид или быть инфильтрированными лейкоцитами и мононуклеарными клетками воспаления.
При электронной микроскопии обнаруживают разрывы гломерулярной базальной мембраны и отложения фибрина. Характерной чертой анти-ГБМ гломерулонефрита является отсутствие отложений иммунных комплексов. При иммунофлюоресцентном исследовании в базальной мембране капиллярных петель клубочков выявляют линейные отложения IgG и С3. Линейные отложения IgG иногда определяются и при других заболеваниях почек (диабетической нефропатии), поэтому требуется дополнительное определение циркулирующих анти-ГБМ антител. При сахарном диабете линейные отложения не являются отражением специфической иммунной реакции и обычно содержат альбумины и фибриноген.
Иммунокомплексный гломерулонефрит с полулуниями встречается в 40 % случаев гломерулонефрита с полулуниями и протекает менее агрессивно, чем остальные формы. Чаще всего он служит осложнением других иммунокомплексных нефритов, например постинфекционного гломерулонефрита, IgA-нефропатии, криоглобулинемического гломерулонефрита, болезни Шенлейна—Геноха, системной красной волчанки. Изредка причину установить не удается, и заболевание считается идиопатическим.
Гистологическая картина иммунокомплексного гломерулонефрита с полулуниями зависит от основного заболевания почек. В участках почечных клубочков, прилегающих к полулуниям, часто обнаруживается некроз, но не такой выраженный, как при анти-ГБМ и малоиммунном гломерулонефритах. Признак, позволяющий дифференцировать иммунокомплексный гломерулонефрит с полулуниями от названных двух вариантов, – наличие утолщения стенок капилляров и пролиферации клеток капилляров почечных клубочков.
При электронной микроскопии и иммунофлюоресценции обнаруживают иммунные комплексы, демонстрирующие гранулярный тип свечения.
Малоиммунный гломерулонефрит с полулуниями наблюдается наиболее часто (в 40–50 % случаев гломерулонефрита с полулуниями). Диагноз устанавливается после исключения участия в патогенезе иммунных комплексов и анти-ГБМ антител. Иммунные комплексы в клубочках при этом обнаруживают очень редко или не обнаруживают вовсе. Малоиммунный гломерулонефрит развивается или самостоятельно, или как компонент системного некротизирующего васкулита (гранулематоза Вегенера, микроскопического полиангиита). У 80–90 % пациентов с малоиммунным гломерулонефритом с полулуниями обнаруживают циркулирующие ANCA, направленные к протеиназе 3 или миелопероксидазе; у остальных пациентов циркулирующие ANCA не выявляются. У ANCA-отрицательных пациентов, как правило, заболевание ограничивается поражением почек, но протекает тяжелее: у них более высокая протеинурия, больше выражен нефротический синдром и хуже прогноз.
Гистологическая картина малоиммунного гломерулонефрита с полулуниями схожа с таковой анти-ГБМ гломерулонефрита. При этом морфологические изменения у ANCA-положительных и ANCA-отрицательных пациентов несколько отличаются. У ANCA-отрицательных пациентов отмечают более выраженные поражения почечных клубочков (меньше сохранных клубочков) и более выраженный интерстициальный фиброз.
При малоиммунном гломерулонефрите в фиброзно-клеточных полулуниях обнаруживают миофибробласты, обладающие одновременно чертами фибробластов и гладкомышечных клеток и способные синтезировать коллаген и таким образом приводить к дальнейшему фиброзированию полулуний. Источником миофибробластов являются, вероятнее всего, париетальные эпителиальные клетки, подвергающиеся эпителиально-мезенхимальной трансдифференцировке под влиянием различных цитокинов и факторов роста. В пользу этой гипотезы о происхождении миофибробластов свидетельствует коэкспрессия ими на переходной стадии эпителиальных маркеров (цитокератинов) и гладкомышечного актина. Этот процесс является довольно ранним в фиброзировании клубочков, и такие «переходные» клетки (в отличие от зрелых миофибробластов) обнаруживают только на стадии клеточных и фиброзно-клеточных полулуний. Другими возможными источниками миофибробластов являются мезангиальные клетки почечных клубочков и перигломерулярные интерстициальные фибробласты, мигрирующие в пространство Боумена через разрывы в капсуле и трансформирующиеся в миофибробласты под действием трансформирующего фактора роста b. Под действием этого фактора роста они синтезируют коллагены III и IV типов.
Таблица 3.1
Этиология быстропрогрессирующего гломерулонефрита
Особенности клинической эволюции и прогноза быстропрогрессирующего гломерулонефрита во многом определяются типом участвующих в почечном поражении антител и вариантом их взаимодействия с гломерулярной базальной мембраной. Именно поэтому общепризнано разделение быстропрогрессирующего гломерулонефрита на иммунопатогенетические варианты (табл. 3.2), определяемые на основании оцениваемого при иммунофлуоресцентной микроскопии типа свечения антител в образцах ткани почки, полученной при биопсии, и наличия определенных сывороточных маркеров (антител к цитоплазме нейтрофилов (ANCA), анти-БМК-антител).
Таблица 3.2
Иммунопатогенетические типы быстропрогрессирующего нефрита
При II (иммунокомплексном) типе депозиты иммунных комплексов обнаруживают в мезангии и стенке капилляров почечного клубочка. Характерные серологические маркеры отсутствуют, за исключением сниженного сывороточного уровня комплемента, который может косвенно указывать на связь быстропрогрессирующего гломерулонефрита с HCV-ассоциированной смешанной криоглобулинемией или системной красной волчанкой.
Почечное поражение при III (малоиммунном) типе быстропрогрессирующего гломерулонефрита определяется клеточными иммунными реакциями, индуцируемыми ANCA. Быстропрогрессирующий гломерулонефрит III типа может быть локально-почечным, или, сочетаясь с поражением других органов, существовать как одно из проявлений системных ANCA-ассоциированных некротизирующих васкулитов (гранулематоз Вегенера, микроскопический полиангиит).
Более половины всех случаев быстропрогрессирующего гломерулонефрита относится к III типу, частота встречаемости I и II типов примерно одинакова (20–25 %).
Одним из центральных звеньев патогенеза быстропрогрессирующего экстракапиллярного гломерулонефрита с полулуниями считают проникновение белков плазмы крови и воспалительных клеток в пространство капсулы Шумлянского—Боумена. Накопление макрофагов вследствие миграции их предшественников из циркулирующей крови и пролиферации резидентных клеток дополняется миграцией из почечного интерстиция фибробластов и миофибробластов. Эти клетки начинают в избытке продуцировать компоненты экстрацеллюлярного матрикса (фибронектин, коллагены I и III типов), что способствует значительному повышению интенсивности фиброза полулуний, лежащего в основе необратимого ухудшения фильтрационной функции почек. Следует тем не менее подчеркнуть, что далеко не всегда при быстропрогрессирующем гломерулонефрите удается установить четкое соответствие между выраженностью почечной недостаточности и количеством полулуний, подвергшихся фибротической трансформации. С одной стороны, суммарное число почечных клубочков, в которых выявляются полулуния, можно считать решающим в развитии быстропрогрессирующего гломерулонефрита. Однако его формирование регистрируют и тогда, когда число клубочковых полулуний не достигает 50 % или они практически отсутствуют.
Межклеточным взаимодействиям в полулуниях, регулируемым определенными хемокинами (моноцитарным хемотаксическим протеином I типа (MCP-1), макрофагальным воспалительным протеином типа 1a (MIP1a)), очевидно, принадлежит решающая роль в формировании быстропрогрессирующего гломерулонефрита. Так, при экстракапиллярном гломерулонефрите установлена прямая корреляция между выраженностью фиброза полулуний и количеством экспрессирующих a-гладкомышечный актин миофибробластов в интерстиции в местах, близких к точкам разрыва капсулы Боумена; там же, а также в интерстициальных клетках, окружающих сосуды, с наибольшей интенсивностью синтезируется коллаген III типа. Трансформирующий фактор роста b-1 при экстракапиллярном гломерулонефрите обнаруживают преимущественно в канальцевых эпителиоцитах. Число миофибробластов, экспрессирующих a-гладкомышечный актин, и количество коллагена IV в почечном интерстиции позволяют предсказать вероятность необратимого ухудшения функции почек и ответ на иммуносупрессивную терапию. Показано, что нарастание экспрессии миофибробластов ассоциировано с усилением процессов апоптоза клеток почечного тубуло-интерстиция с одновременным увеличением синтеза медиаторов фиброгенеза, в первую очередь трансформирующего фактора роста-b.
Установлено, что при экстракапиллярном гломерулонефрите интенсивная экспрессия трансформирующего фактора роста-b ассоциирована с увеличением числа полулуний, подвергшихся фибротической трансформации. Более того, мочевая экскреция трансформирующего фактора роста-b у больных экстракапиллярным гломерулонефритом, не ответивших на иммуносупрессивную терапию и продемонстрировавших быстрое развитие терминальной почечной недостаточности, не менее чем в 2,5 раза превосходит таковую у тех пациентов, у кого прогрессирование заболевания удалось задержать с помощью иммуносупрессивной терапии.
Роль избытка трансформирующего фактора роста-b в прогрессировании быстропрогрессирующего экстракапиллярного гломерулонефрита, очевидно, не исчерпывается только его участием в процессах почечного фиброгенеза. Хорошо известно, что трансформирующий фактор роста-b – один из наиболее мощных физиологических индукторов клеточного апоптоза. Высокая интенсивность синтеза этого медиатора при экстракапиллярном гломерулонефрите может отражать блокаду соответствующих рецепторов и связанное с ней торможение процессов апоптоза, представляющее собой один из механизмов формирования полулуний. В норме трансформирующий фактор роста-b индуцирует клеточный апоптоз путем взаимодействия со специфическим рецептором BMRP (bone morphogenetic protein receptor – рецептором костного морфогенетического белка). Установлено, что при малоиммунном (тип III) варианте быстропрогрессирующего гломерулонефрита в почечных клубочках и интерстиции, в том числе в области полулуний, существенно возрастает экспрессия белка гремлина – антагониста BMRP, препятствующего таким образом взаимодействию этого белкового рецептора с трансформирующим фактором роста-b. В результате блокады BMRP гремлином миофибробласты и фибробласты не вступают в процесс апоптоза, а продолжают пролиферировать и дифференцироваться, что приводит к дальнейшему образованию новых полулуний и их интенсивному фиброзу, несмотря на возникающую по механизму отрицательной обратной связи гиперпродукцию трансформирующего фактора роста-b, который в этой ситуации начинает реализовывать свое профиброгенное действие другими путями, например через провокацию экспрессии медиаторов эндотелий-зависимого звена гемостаза (ингибитора активатора плазминогена-1).
Представление о быстропрогрессирующем гломерулонефрите как о заболевании, в основе развития которого лежит патология индуцируемого трансформирующим фактором роста-b апоптоза, открывает новые перспективы в лечении этого заболевания. Наряду с ингибированием блокирующего рецепторы BMRP гремлина можно рассчитывать на терапевтическую эффективность введения в почечную ткань доноров BMRP, в том числе клеток костного мозга. Показано, что костный мозг может быть источником почечных мезангиоцитов и эндотелиоцитов, трансдифференциация которых осуществляется в условиях характерного микроокружения и при участии некоторых факторов роста, в частности тромбоцитарного. Гиперпродукция блокаторов BMRP или недостаточная экспрессия этих рецепторов могут рассматриваться как одно из наиболее важных обстоятельств, предрасполагающих к развитию именно быстропрогрессирующего экстракапиллярного гломерулонефрита. Более того, сама по себе недостаточность рецепторов BMRP может быть генетически детерминированной (аналогично первичной легочной гипертензии – другому заболеванию, в основе которого лежит патология апоптоза отдельных клеточных пулов с их избыточной пролиферацией, при котором носительство мутантных форм гена BMRP четко продемонстрировано и обусловливает семейные случаи). Следовательно, изучение вариантов гена BMRP и интенсивности экспрессии его блокаторов (в частности, гремлина) может способствовать более детальному пониманию процессов, лежащих в основе развития быстропрогрессирующего гломерулонефрита, в том числе объяснению его развития у лиц, исходно страдающих другим хроническим заболеванием почек, например диабетической нефропатией.
Понятно, что каждый из типов быстропрогрессирующего гломерулонефрита имеет определенные патогенетические особенности.
Развитие синдрома Гудпасчера (тип I – антительный – быстропрогрессирующего гломерулонефрита) связывают со специфическими анти-БМК антителами, связывающимися с базальной мембраной альвеол и почечного клубочка и обладающими высокой специфичностью к a-3 цепи коллагена IV типа. Cвязь анти-БМК антител с a-3 цепью коллагена IV типа происходит через содержащийся в ней так называемый неколлагеновый (NC) домен. Cуществует 4 типа анти-БМК антител (А, В, АВ, Х). А и В типы взаимодействуют с определенными эпитопами (А или В, соответственно), АВ – с обоими названными эпитопами, точка приложения действия анти-БМК антител Х типа неясна. Все эти эпитопы расположены на NC домене.
В настоящее время синдром Гудпасчера продолжают интенсивно изучать на экспериментальных моделях: создан синтетический аналог соответствующего антигена, при введении которого животным у них развивается легочно-почечный синдром, обязательно включающий антительный быстропрогрессирующий гломерулонефрит. У крыс с синдромом Гудпасчера удалось детально проследить эволюцию почечного поражения. Депозиты IgG на базальной мембране формируются через 2 недели после иммунизации, одновременно констатируют отек клубочковых эндотелиоцитов и появление альбуминурии. Клеточный инфильтрат представлен преимущественно СD4-позитивными Т-лимфоцитами. На 3-й неделе начинают образовываться полулуния, в капсуле Боумена накапливается большое количество фибрина, гломерулярный инфильтрат представлен преимущественно CD8-позитивными Т-лимфоцитами и макрофагами. Характерны множественные разрывы базальной мембраны.
Показано, что в формировании тканевого повреждения при синдроме Гудпасчера существенную роль играют Т-лимфоциты. Продемонстрировано значительное нарастание числа CD4-позитивных Т-лимфоцитов, распознающих NC-домен a-3 цепи коллагена IV типа в периферической крови в острую фазу заболевания. Если обострение удается прервать, число их существенно уменьшается. Развитие синдрома Гудпасчера может быть также обусловлено нарушениями регуляции экспрессии IgG. Так, в эксперименте удавалось воспроизвести синдром Гудпасчера у мышей с отсутствующим Fc-гамма рецептором IIB, играющим центральную роль в контроле экспрессии IgG. Часть случаев синдрома Гудпасчера может также быть обусловлена моноклональной пролиферацией IgA.
При синдроме Гудпасчера фибробласты почечного тубуло-интерстиция активно экспрессируют a-гладкомышечный актин, в то время как виментина и трансформирующего фактора роста-b на их поверхности обнаружить не удается. Показано, что блокада моноцитарного хемоаттрактантного протеина I типа позволяет уменьшить выраженность тубуло-интерстициального повреждения при синдроме Гудпасчера, что также открывает определенные перспективы в лечении данного заболевания.
Иммунокомплексный (тип II) вариант быстропрогрессирующего гломерулонефрита развивается при наличии соответствующего субстрата для образования иммунных комплексов. В качестве такового могут выступать бактериальные антигены, лекарственные препараты или их метаболиты, криоглобулины при HCV-ассоциированной смешанной криоглобулинемии, антитела к ДНК при системной красной волчанке.
Почечное поражение при III (малоиммунном, pauci) типе быстропрогрессирующего гломерулонефрита определяется тканьдеструктивным действием ANCA, степень повышения сывороточной концентрации которых, как правило, отражает активность заболевания. Взаимодействие между Fc-рецепторами нейтрофилов, миелопероксидазой, протеинкиназой-3 и ANCA сопровождается гиперпродукцией супероксид-иона, перекисей, а также провоспалительных цитокинов. Одновременно возрастает экспрессия b2-интегрина – молекулы адгезии, принимающей непосредственное участие во взаимодействии нейтрофилов с эндотелиоцитами (повреждение эндотелиоцитов при ANCA-ассоциированном быстропрогрессирующем гломерулонефрите может также быть связано с продукцией антиэндотелиальных антител). В тканях-мишенях ANCA формируются также олигоклональные пулы Т-лимфоцитов. Эти Т-лимфоциты, модулируя продукцию интерферона-g и a-фактора некроза опухолей, принимают непосредственное участие в образовании полулуний. Более тяжелое почечное поражение наблюдают при преобладании CD28– Т-лимфоцитов.
Детали патогенеза отдельных вариантов быстропрогрессирующего гломерулонефрита продолжают уточняться. Для разработки селективных терапевтических воздействий необходимо уточнение механизмов формирования полулуний и индукторов их фибротической трансформации.
Морфология
Гломерулонефрит с полулуниями (быстропрогрессирующий, подострый, злокачественный, экстракапиллярный пролиферативный) – тяжелая форма гломерулонефрита, при которой в большинстве клубочков формируются эпителиальные полулуния.
Важный патогенетический фактор формирования полулуний в пространстве Боумена – выход фибрина в просвет капсулы почечного тельца. Вероятно, фибрин и другие белки плазмы крови, а также моноциты попадают в просвет капсулы почечного тельца через разрывы в воспаленных капиллярах клубочков. Фибрин и его компоненты являются стимулом для миграции моноцитов. Макрофаги, образующиеся из моноцитов, пролиферирующие париетальные эпителиальные клетки и полиморфно-ядерные лейкоциты образуют «клеточные» полулуния. Эти полулуния постепенно сдавливают капиллярный клубочек, серьезно затрудняя и даже прекращая образование первичной мочи с развитием олигоурии и анурии. Спавшиеся капиллярные петли пропитываются фибрином. По мере прогрессирования процесса полулуния приобретают фиброзный характер, в них появляются фибробласты и коллаген. В финале клубочек полностью склерозируется.
Гломерулонефрит с полулуниями в большой степени является результатом клеточно-опосредованного иммунного повреждения клубочков. В развитии данного заболевания определенная роль отводится гиперчувствительности замедленного типа (ГЗТ), при которой сенсибилизированные Т-лимфоциты привлекают и активируют макрофаги. При этом отмечается значительная локальная экспрессия тканевого фактора в почечных клубочках. Наблюдается выраженная гломерулярная аккумуляция CD4+ Т-лимфоцитов. Обнаружение CD8+ Т-лимфоцитов свидетельствует об участии Т-клеточно-опосредованной цитотоксичности в патогенезе гломерулонефрита с полулуниями. Цитотоксические Т-лимфоциты вызывают повреждение как секретируя цитотоксические молекулы, так и с помощью клеточных медиаторов (фактора некроза опухолей a-ФНО и Fas-лиганды), взаимодействующих со своими рецепторами на клетках-мишенях и вызывающих их апоптоз. Известно, что в интрагломерулярных мононуклеарных клетках происходит усиленная продукция РНК провоспалительных цитокинов ФНО-a и интерлейкина-1b (ИЛ-1b). Считается, что ИЛ-1b продуцируют в основном макрофаги в полулуниях, в то время как ФНОa продуцируется клетками почечных клубочков, в том числе мезангиальных, и эпителиальными клетками почечных канальцев. Продукция ИЛ-1b и его взаимодействие со своим рецептором I типа на клетках почечных клубочков вызывает экспрессию ФНО-a и дальнейшее повреждение клубочков.
Важную роль в регуляции макрофагальной инфильтрации почечных клубочков при гломерулонефрите с полулуниями играет моноцитарный хемоаттрактантный белок-1 (MCP-1) и его рецепторы. В почечных клубочках отмечается усиленная экспрессия MCP-1 клетками полулуний, париетальными эпителиальными клетками, эпителием канальцев и инфильтрирующими интерстиций лейкоцитами. Также возрастает экспрессия хемокинового рецептора 2В, обнаруживаемого в основном на мононуклеарных лейкоцитах, инфильтрирующих клубочек и полулуния. В привлечении лейкоцитов и формировании инфильтрата определенную роль играют адгезивные молекулы ICAM-1 и VCAM-1, повышенная экспрессия которых выявляется в пораженных почечных клубочках пациентов с гломерулонефритом с полулуниями (анти-ГБМ и малоиммунном типах).
Макроскопически при быстропрогрессирующем гломерулонефрите почки увеличены в размерах, дряблые; кора широкая, желто-серая, тусклая с красным крапом, резко отграничена от темно-красных пирамид – «большая пестрая почка», или красная, сливается с пирамидами – «большая красная почка».
В гистологической картине доминирует образование полулуний, которые могут занимать только отдельные сегменты пространства капсулы почечного тельца или полностью окружать клубочек. Развитие полулуний проходит несколько стадий: от клеточных полулуний к фиброзно-клеточным, а затем – фиброзным. Как уже было сказано, «клеточные» полулуния, занимающие пространство между капсулой и капиллярным клубочком, представлены пролиферирующими париетальными эпителиальными клетками и макрофагами, также в них обнаруживаются нейтрофилы и лимфоциты. В течение нескольких недель в полулуниях развиваются процессы организации с формированием соединительнотканного матрикса, который хорошо выявляется при серебрении. Полулуния, полностью окружающие клубочки, вызывают тотальный склероз. Наблюдаются также отек, инфильтрация и склероз стромы, атрофия канальцев.
Быстропрогрессирующий гломерулонефрит подразделяют на 3 группы: анти-ГБМ гломерулонефрит; иммунокомплексный гломерулонефрит с полулуниями; малоиммунный гломерулонефрит с полулуниями.
Анти-ГБМ гломерулонефрит характеризуется линейными отложениями IgG и С3 компонента комплемента вдоль гломерулярной базальной мембраны. Вызывается аутоантителами к a-3 цепи коллагена IV типа. Составляет от 10 до 20 % случаев гломерулонефрита с полулуниями. Анти-ГБМ гломерулонефрит может ограничиваться поражением почек или развиваться в форме почечно-легочного синдрома. Классическим примером последнего является быстропрогрессирующий гломерулонефрит, связанный с синдромом Гудпасчера. Антитела к БМК перекрестно реагируют с базальными мембранами легочных альвеол. Обусловливают появление клинической картины легочных кровоизлияний, сопровождающихся почечной недостаточностью. Кроме того, анти-ГБМ гломерулонефрит может быть осложнением других заболеваний, например развиваться de novo после трансплантации почки у пациентов с синдромом Альпорта, а также обнаруживаться у пациентов с мембранозным гломерулонефритом с образованием полулуний. Примерно у 1/3 пациентов с анти-ГБМ гломерулонефритом выявляются циркулирующие антинейтрофильные цитоплазматические антитела (ANCA), и данное заболевание у них ассоциировано с васкулитом мелких сосудов различных органов (не только почек и легких).
Типичной картиной при световой микроскопии является некротизирующий гломерулонефрит с очаговым или диффузным формированием полулуний. В пораженных участках капилляров почечных клубочков обнаруживаются скопления лейкоцитов. Непораженные участки могут иметь нормальный вид или быть инфильтрированными лейкоцитами и мононуклеарными клетками воспаления.
При электронной микроскопии обнаруживают разрывы гломерулярной базальной мембраны и отложения фибрина. Характерной чертой анти-ГБМ гломерулонефрита является отсутствие отложений иммунных комплексов. При иммунофлюоресцентном исследовании в базальной мембране капиллярных петель клубочков выявляют линейные отложения IgG и С3. Линейные отложения IgG иногда определяются и при других заболеваниях почек (диабетической нефропатии), поэтому требуется дополнительное определение циркулирующих анти-ГБМ антител. При сахарном диабете линейные отложения не являются отражением специфической иммунной реакции и обычно содержат альбумины и фибриноген.
Иммунокомплексный гломерулонефрит с полулуниями встречается в 40 % случаев гломерулонефрита с полулуниями и протекает менее агрессивно, чем остальные формы. Чаще всего он служит осложнением других иммунокомплексных нефритов, например постинфекционного гломерулонефрита, IgA-нефропатии, криоглобулинемического гломерулонефрита, болезни Шенлейна—Геноха, системной красной волчанки. Изредка причину установить не удается, и заболевание считается идиопатическим.
Гистологическая картина иммунокомплексного гломерулонефрита с полулуниями зависит от основного заболевания почек. В участках почечных клубочков, прилегающих к полулуниям, часто обнаруживается некроз, но не такой выраженный, как при анти-ГБМ и малоиммунном гломерулонефритах. Признак, позволяющий дифференцировать иммунокомплексный гломерулонефрит с полулуниями от названных двух вариантов, – наличие утолщения стенок капилляров и пролиферации клеток капилляров почечных клубочков.
При электронной микроскопии и иммунофлюоресценции обнаруживают иммунные комплексы, демонстрирующие гранулярный тип свечения.
Малоиммунный гломерулонефрит с полулуниями наблюдается наиболее часто (в 40–50 % случаев гломерулонефрита с полулуниями). Диагноз устанавливается после исключения участия в патогенезе иммунных комплексов и анти-ГБМ антител. Иммунные комплексы в клубочках при этом обнаруживают очень редко или не обнаруживают вовсе. Малоиммунный гломерулонефрит развивается или самостоятельно, или как компонент системного некротизирующего васкулита (гранулематоза Вегенера, микроскопического полиангиита). У 80–90 % пациентов с малоиммунным гломерулонефритом с полулуниями обнаруживают циркулирующие ANCA, направленные к протеиназе 3 или миелопероксидазе; у остальных пациентов циркулирующие ANCA не выявляются. У ANCA-отрицательных пациентов, как правило, заболевание ограничивается поражением почек, но протекает тяжелее: у них более высокая протеинурия, больше выражен нефротический синдром и хуже прогноз.
Гистологическая картина малоиммунного гломерулонефрита с полулуниями схожа с таковой анти-ГБМ гломерулонефрита. При этом морфологические изменения у ANCA-положительных и ANCA-отрицательных пациентов несколько отличаются. У ANCA-отрицательных пациентов отмечают более выраженные поражения почечных клубочков (меньше сохранных клубочков) и более выраженный интерстициальный фиброз.
При малоиммунном гломерулонефрите в фиброзно-клеточных полулуниях обнаруживают миофибробласты, обладающие одновременно чертами фибробластов и гладкомышечных клеток и способные синтезировать коллаген и таким образом приводить к дальнейшему фиброзированию полулуний. Источником миофибробластов являются, вероятнее всего, париетальные эпителиальные клетки, подвергающиеся эпителиально-мезенхимальной трансдифференцировке под влиянием различных цитокинов и факторов роста. В пользу этой гипотезы о происхождении миофибробластов свидетельствует коэкспрессия ими на переходной стадии эпителиальных маркеров (цитокератинов) и гладкомышечного актина. Этот процесс является довольно ранним в фиброзировании клубочков, и такие «переходные» клетки (в отличие от зрелых миофибробластов) обнаруживают только на стадии клеточных и фиброзно-клеточных полулуний. Другими возможными источниками миофибробластов являются мезангиальные клетки почечных клубочков и перигломерулярные интерстициальные фибробласты, мигрирующие в пространство Боумена через разрывы в капсуле и трансформирующиеся в миофибробласты под действием трансформирующего фактора роста b. Под действием этого фактора роста они синтезируют коллагены III и IV типов.