По всем этим причинам свертывание крови затормаживается и не переходит в обычных условиях из локального процесса во всеобщую коагуляцию циркулирующего фибриногена.
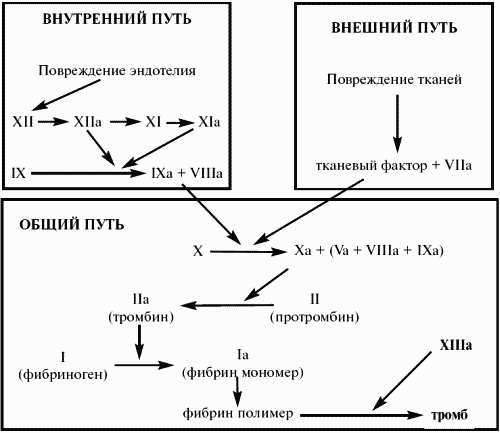
Также в активации начальных этапов свертывания участвуют компоненты калликреин-кининовой системы. Стимуляторами являются фактор ХIIа и его фрагменты, образующиеся в результате расщепления фактора XII калликреином. Комплекс фактор ХIIа – калликреин – высокомолекулярный кининоген (ВМК) ускоряет активацию не только фактора XI, но и фактора VII, реализуя взаимосвязь между внутренним и внешним механизмами свертывания. Еще более важно активирующее влияние тромбопластина и фактора VII на фактор IX, в результате чего даже малые дозы тканевого тромбопластина запускают процесс свертывания крови не только по внешнему, но и по внутреннему пути, через фактор IX. Установлено также, что фактор ХIIа и его фрагменты через калликреин-кининовую систему, а отчасти и непосредственно активируют ряд других плазменных ферментных систем, в том числе фибринолитическую и систему комплемента.
Фактор VIII – многокомпонентная система, состоящая из нескольких субъединиц, участвующих в формировании его коагуляционной активности (VIII : С) и в тромбоцитарно-сосудистом взаимодействии (фактор Виллебранда VIII : FW). Эти субъединицы отличаются разной стабильностью, гетерогенны по генетическому контролю и антигенным свойствам.
Противосвертывающие механизмы
Противосвертывающие механизмы играют ведущую роль в поддержании жидкого состояния крови и в ограничении процесса тромбообразования. Однако они изучены значительно меньше, чем процесс свертывания крови, в связи с чем вопросы функции и физиологической регуляции антикоагулянтного звена системы гемостаза во многом остаются дискуссионными.
Все образующиеся в организме антикоагулянты можно разделить на 2 группы:
Из всех предшествующих антикоагулянтов наиболее активен, универсален по действию и важен для поддержания жидкого состояния крови антитромбин III (AT III). Этот белок, содержащийся в плазме в количестве около 0,3–0,42 г/л, или 3,0–4,7 ммоль/л, инактивирует не только тромбин, но и все другие активированные ферментные факторы свертывания: ХIIа, XIa, IXa, Ха. Он же является плазменным кофактором гепарина – без AT III гепарин почти совершенно не оказывает антикоагулянтного действия. Дефицит AT III, наследственный или вторичный (симптоматический), закономерно приводит к развитию тяжелейшего, часто несовместимого с жизнью тромбоэмболического синдрома. Дефицит всех других предшествующих физиологических антикоагулянтов по раздельности либо в совокупности не создает подобных критических ситуаций. Все вышеперечисленные данные закрепили за AT III репутацию главного ингибитора и модулятора системы свертывания крови. Местом синтеза AT III долгое время считали печень, однако исследования последних лет, в том числе выполненные на клеточных культурах иммунологическими методами, показали, что этот антикоагулянт продуцируется сосудистым эндотелием. Все факторы свертывания AT III инактивирует, образуя с ними эквимолярные комплексные соединения. Гепарин, соединяясь с AT III, резко ускоряет это взаимодействие и фиксирует антикоагулянт на поверхности эндотелиальных клеток, чем повышается тромбоустойчивость внутренней стенки сосудов.
Альфа2-макроглобулин является слабым ингибитором тромбина, действие которого становится ощутимым лишь при депрессии AT III. На долю этого антикоагулянта приходится, по разным авторам, от 4 до 21% антитромбиновой активности дефибринированной плазмы. Несколько больше роль α2-макроглобулина в связывании плазмина, но и в этом случае его действие становится ощутимым после удаления быстродействующего антиплазмина. В отличие от AT III он из всех активированных факторов свертывания взаимодействует только с тромбином. Наследственный дефицит α2-макроглобулина не сопровождается ни сколько-нибудь заметной тромбогенностью, ни существенными сдвигами в свертывающей системе крови, что говорит о его весьма ограниченном значении в регуляции гемостатического потенциала крови.
Более выражено ингибирующее действие на тромбин и другие активированные факторы свертывания (IXa, XIa и ХПа) α1-антитрипсина и ингибитора 1 компонента комплемента. Однако и при их дефиците не наблюдается значительных нарушений гемостаза, что, очевидно, связано с одинаково выраженным ослаблением инактивации как свертывания крови, так и фибринолиза, вследствие чего сохраняется динамическое равновесие между этими системами.
Антикоагулянты, образующиеся в процессе свертывания крови и фибринолиза. Многие прокоагулянты и их метаболиты в процессе свертывания крови и фибринолиза приобретают антикоагулянтные свойства. Так, фибрин адсорбирует и инактивирует образующийся при свертывании тромбин, вследствие чего фибрин обозначается как антитромбин I. Эта инактивация настолько велика, что в сыворотке, как известно, остаются ничтожно малые количества тромбина. Имеются указания, что фибринопептиды, отщепляемые от фибриногена тромбином, также обладают антикоагулянтным действием.
Самоторможение наблюдается и на других этапах свертывания. Так, тромбин действует ферментативно на протромбин, отщепляя от него ингибитор фактора Ха; фактор Va после участия в свертывании начинает тормозить превращение протромбина в тромбин, а фактор ХIа после взаимодействия с фактором XII начинает тормозить его дальнейшую активацию. Мощные антикоагулянты, обладающие антитромбиновым и антиполимеразным действием, образуются в процессе фибринолиза.
Все вышеперечисленные данные в очередной раз свидетельствуют о том, что в свертывающей системе крови на всех этапах каскада действуют силы самоограничения процесса, одни и те же факторы могут выступать вначале как коагулянты, а затем – как антикоагулянты.
Схема 2 Система свертывания крови
Фибринолитическая (плазминовая) система
Часть II. Частная гематология
Глава 1. Анемии
Острая постгеморрагическая анемия
Железодефицитные анемии
Также в активации начальных этапов свертывания участвуют компоненты калликреин-кининовой системы. Стимуляторами являются фактор ХIIа и его фрагменты, образующиеся в результате расщепления фактора XII калликреином. Комплекс фактор ХIIа – калликреин – высокомолекулярный кининоген (ВМК) ускоряет активацию не только фактора XI, но и фактора VII, реализуя взаимосвязь между внутренним и внешним механизмами свертывания. Еще более важно активирующее влияние тромбопластина и фактора VII на фактор IX, в результате чего даже малые дозы тканевого тромбопластина запускают процесс свертывания крови не только по внешнему, но и по внутреннему пути, через фактор IX. Установлено также, что фактор ХIIа и его фрагменты через калликреин-кининовую систему, а отчасти и непосредственно активируют ряд других плазменных ферментных систем, в том числе фибринолитическую и систему комплемента.
Фактор VIII – многокомпонентная система, состоящая из нескольких субъединиц, участвующих в формировании его коагуляционной активности (VIII : С) и в тромбоцитарно-сосудистом взаимодействии (фактор Виллебранда VIII : FW). Эти субъединицы отличаются разной стабильностью, гетерогенны по генетическому контролю и антигенным свойствам.
Противосвертывающие механизмы
Противосвертывающие механизмы играют ведущую роль в поддержании жидкого состояния крови и в ограничении процесса тромбообразования. Однако они изучены значительно меньше, чем процесс свертывания крови, в связи с чем вопросы функции и физиологической регуляции антикоагулянтного звена системы гемостаза во многом остаются дискуссионными.
Все образующиеся в организме антикоагулянты можно разделить на 2 группы:
1) существующие или синтезируемые самостоятельно, возникающие независимо от свертывания крови, фибринолиза или активации других ферментных систем;Физиологические антикоагулянты, образующиеся независимо от протеолиза. К ним относятся белковые и фосфолипидные ингибиторы начальной фазы свертывания крови, из которых наиболее активен и физиологически важен относящийся к α2-глобулинам ингибитор фактора XIа. Слабее действует на начальные фазы свертывания липидный антикоагулянт.
2) образующиеся в процессе протеолиза – свертывания крови, фибринолиза. Химически антикоагулянты относятся к разным группам веществ – белкам, пептидам, липидам, мукополисахаридам.
Из всех предшествующих антикоагулянтов наиболее активен, универсален по действию и важен для поддержания жидкого состояния крови антитромбин III (AT III). Этот белок, содержащийся в плазме в количестве около 0,3–0,42 г/л, или 3,0–4,7 ммоль/л, инактивирует не только тромбин, но и все другие активированные ферментные факторы свертывания: ХIIа, XIa, IXa, Ха. Он же является плазменным кофактором гепарина – без AT III гепарин почти совершенно не оказывает антикоагулянтного действия. Дефицит AT III, наследственный или вторичный (симптоматический), закономерно приводит к развитию тяжелейшего, часто несовместимого с жизнью тромбоэмболического синдрома. Дефицит всех других предшествующих физиологических антикоагулянтов по раздельности либо в совокупности не создает подобных критических ситуаций. Все вышеперечисленные данные закрепили за AT III репутацию главного ингибитора и модулятора системы свертывания крови. Местом синтеза AT III долгое время считали печень, однако исследования последних лет, в том числе выполненные на клеточных культурах иммунологическими методами, показали, что этот антикоагулянт продуцируется сосудистым эндотелием. Все факторы свертывания AT III инактивирует, образуя с ними эквимолярные комплексные соединения. Гепарин, соединяясь с AT III, резко ускоряет это взаимодействие и фиксирует антикоагулянт на поверхности эндотелиальных клеток, чем повышается тромбоустойчивость внутренней стенки сосудов.
Альфа2-макроглобулин является слабым ингибитором тромбина, действие которого становится ощутимым лишь при депрессии AT III. На долю этого антикоагулянта приходится, по разным авторам, от 4 до 21% антитромбиновой активности дефибринированной плазмы. Несколько больше роль α2-макроглобулина в связывании плазмина, но и в этом случае его действие становится ощутимым после удаления быстродействующего антиплазмина. В отличие от AT III он из всех активированных факторов свертывания взаимодействует только с тромбином. Наследственный дефицит α2-макроглобулина не сопровождается ни сколько-нибудь заметной тромбогенностью, ни существенными сдвигами в свертывающей системе крови, что говорит о его весьма ограниченном значении в регуляции гемостатического потенциала крови.
Более выражено ингибирующее действие на тромбин и другие активированные факторы свертывания (IXa, XIa и ХПа) α1-антитрипсина и ингибитора 1 компонента комплемента. Однако и при их дефиците не наблюдается значительных нарушений гемостаза, что, очевидно, связано с одинаково выраженным ослаблением инактивации как свертывания крови, так и фибринолиза, вследствие чего сохраняется динамическое равновесие между этими системами.
Антикоагулянты, образующиеся в процессе свертывания крови и фибринолиза. Многие прокоагулянты и их метаболиты в процессе свертывания крови и фибринолиза приобретают антикоагулянтные свойства. Так, фибрин адсорбирует и инактивирует образующийся при свертывании тромбин, вследствие чего фибрин обозначается как антитромбин I. Эта инактивация настолько велика, что в сыворотке, как известно, остаются ничтожно малые количества тромбина. Имеются указания, что фибринопептиды, отщепляемые от фибриногена тромбином, также обладают антикоагулянтным действием.
Самоторможение наблюдается и на других этапах свертывания. Так, тромбин действует ферментативно на протромбин, отщепляя от него ингибитор фактора Ха; фактор Va после участия в свертывании начинает тормозить превращение протромбина в тромбин, а фактор ХIа после взаимодействия с фактором XII начинает тормозить его дальнейшую активацию. Мощные антикоагулянты, обладающие антитромбиновым и антиполимеразным действием, образуются в процессе фибринолиза.
Все вышеперечисленные данные в очередной раз свидетельствуют о том, что в свертывающей системе крови на всех этапах каскада действуют силы самоограничения процесса, одни и те же факторы могут выступать вначале как коагулянты, а затем – как антикоагулянты.
Схема 2 Система свертывания крови
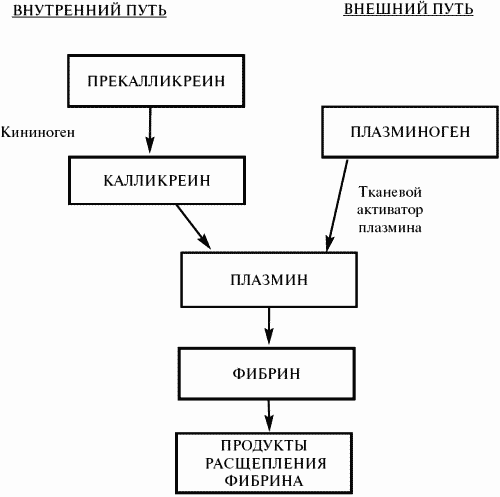
Фибринолитическая (плазминовая) система
Ферментная система, обеспечивающая растворение фибрина в кровяном русле, получила название фибринолитической, или плазминовой, системы. Это растворение осуществляется основным компонентом указанной системы – фибринолизином (или плазмином), который в плазме содержится в виде профермента (плазминогена) в концентрации около 20,6 + 3,6 мг%. Как в плазме, так и в тканях плазминоген содержится в виде двух или более молекулярных форм, отличающихся друг от друга способами выделения, особенностями активации и инактивации. Каждая из двух основных форм состоит из нескольких молекулярных подформ:
1) нативный плазминоген с NH2-терминальной глютаминовой кислотой – «глю-плазминоген»;
2) подвергшийся ограниченному протеолизу плазминоген с NH2-терминальным лизином, валином или метионином – «лиз-плазминоген». Лиз-плазминоген в 10–20 раз быстрее трансформируется активаторами в плазмин, имеет значительно более выраженное, чем глю-плазминоген, сродство к фибрину и значительно быстрее последнего метаболизируется – его Т1/2 в циркуляции около 0,8 суток, а глю-плазминогена – 1,24 ± 0,29 суток. По механизму протеолитического действия плазмин наиболее близок к трипсину.
После активации плазминоген быстро исчезает из кровотока – блокируется антиплазминами и удаляется. Вслед за введением больших доз стрептокиназы или урокиназы уровень плазминогена в крови снижается до нуля, но затем в течение 12–28 ч восстанавливается, если прекращена его дальнейшая активация.
Эта способность активаторов фибринолиза быстро истощать запасы плазминогена в крови и на время оставлять больного без ферментативного фибринолиза важна для клиники и должна учитываться при лечении тромбозов и синдрома диссеминированного внутрисосудистого свертывания крови.
Существующие в организме механизмы активации плазминогена весьма разнообразны, но, подобно механизмам свертывания крови, они также могут быть подразделены на две основные группы – с внутренней и внешней активацией.
Ведущий внутренний механизм запускается теми же факторами, какие инициируют свертывание крови, а именно фактором ХIIа, который, взаимодействуя с прекалликреином и высокомолекулярным кининогеном плазмы (ВМК), активирует плазминоген. Такой путь фибринолиза – базисный, обеспечивающий активацию плазминовой системы не вслед за свертыванием крови, а одновременно с ним. Он функционирует по «замкнутому циклу», поскольку образующиеся первые порции калликреина и плазмина вызывают протеолиз фактора XII, отцепляя фрагменты, под действием которых нарастает изменение прекалликреина в калликреин. Такая интенсивная самоактивация приводит к тому, что ХIIа-калликреин-зависимый фибринолиз при интенсивном внутрисосудистом свертывании крови истощается быстро, раньше других механизмов фибринолиза.
Лимитирующими факторами являются в первую очередь ВМК и прекалликреин. Их плазменный резерв быстро истощается, тогда как уровень плазминогена остается в крови еще достаточно высоким. В таких условиях ХIIа-зависимый фибринолиз уже не функционирует, но поддается другим (не калликреиновым) способам активации – стрептокиназой и урокиназой. Лишь вслед за этим возможно истощение запасов плазминогена, что делает неэффективным любые способы активации плазминовой системы. Определенное участие в активации внутреннего механизма фибринолиза принимает, по-видимому, и фактор Виллебранда. В частности, на образцах плазмы с дефицитом ВМК показано, что фактор Виллебранда в 2–3 раза усиливает превращение прекалликреина в калликреин под влиянием фрагментов фактора XII. В присутствии ВМК, обладающего значительно более мощным влиянием на активацию прекалликреина, это действие фактора Виллебранда становится малоощутимым.
Заслуживает внимания то обстоятельство, что если в свертывании крови компонентам калликреин-кининовой системы отводится в определенной мере вспомогательная функция, то в гуморальном механизме фибринолиза это один из ведущих механизмов. Возможно, именно поэтому при генетически обусловленном дефиците плазменного прекалликреина (дефект Флетчера) или ВМК (дефект Фитцжеральда – Вильсона) у больных нет кровоточивости и вместе с тем прослеживается наклонность к тромбозам.
Важнейшими стимуляторами внешнего механизма фибринолиза являются белковые активаторы плазминогена, синтезируемые в сосудистой стенке. Эти активаторы подразделяются на высокомолекулярные и низкомолекулярные фракции, обнаруживают высокое сродство к фибрину. Физиологическая регуляция синтеза и выделения в кровь сосудистых активаторов изучена недостаточно. Тем не менее известно, что их интенсивный выброс происходит при нарушении проходимости сосудов, в том числе и при пережатии сосуда манжетой, а также при физических нагрузках, под влиянием вазоактивных веществ. Определение эуглобулинового лизиса до и после пережатия сосуда (манжеточная проба) используется для оценки резерва сосудистых активаторов плазминогена и функциональной полноценности механизмов их либерации. Депрессия данных механизмов характерна для ряда тромбофилических состояний. Стероидные гормоны анаболического действия повышают синтез в эндотелии активаторов фибринолиза, с чем отчасти связывается их благоприятное влияние на течение флеботромботической болезни.
Мощные активаторы плазминогена содержатся также в клетках крови – эритроцитах, тромбоцитах и особенно лейкоцитах. При внутрисосудистом свертывании крови, тромбообразовании, воздействии эндотоксином, активации системы комплемента, гемолизе эти активаторы освобождаются из клеток в «плазматическую атмосферу» и активируют плазминоген.
Более того, установлено, что гранулоциты секретируют не только активатор плазминогена, но и внутриклеточные протеазы (цитокиназы), которые самостоятельно, без участия плазмина, переваривают фибрин. При этом образуются иные продукты расщепления фибрина, чем при его плазминовом расщеплении.
Следовательно, лейкоциты обеспечивают функционирование самостоятельного (неплазминового) механизма растворения фибрина. Этот альтернативный механизм играет важную роль в ограничении размеров тромбов и в деблокировании микроциркуляторного русла при диссеминированном внутрисосудистом свертывании крови.
Разнообразные активаторы плазминогена (цитокиназы) содержатся и в других тканях и клетках, особенно в эпителиальной, мышечной и мезенхимальной, а также в секретах и экскретах – моче, молоке, желчи, слюне. Некоторые из них поступают в определенных количествах в кровь, участвуя в активации плазминогена. В частности, таким свойством обладает урокиназа – активатор фибринолиза, синтезируемый в почечном эпителии и выделяющийся с мочой. В кровь поступает небольшое количество урокиназы, ответственное приблизительно за 10–15% общей плазминоген-активаторной функции. В настоящее время установлено, что большинство тканевых активаторов плазминоген идентично сосудистому, эндотелиальному.
Фибринолиз ингибируется рядом антиактиваторов и антиплазминов, из которых наиболее важен недавно открытый быстродействующий антиплазмин, относящийся к α2-глобулинам (молекулярная масса 65 000–70 000) и содержащийся в плазме в количестве 70 мг/л. Этого количества достаточно, чтобы нейтрализовать более 2/3 всего плазмина, образующегося при максимальной активации плазминогена. Однако плазмин, связанный с фибрином, хуже комплексируется с антиплазмином, чем при циркуляции в свободном состоянии. Антиплазмин ослабляет процесс связывания плазминогена с фибрином. Присутствие в плазме циркулирующих комплексов плазмин – антиплазмин, как и комплексов тромбин-ATIII, служит признаком интенсивного внутрисосудистого свертывания крови и активации фибринолиза. Выявление этих комплексов облегчается тем, что в них появляются новые антигенные свойства (так называемые неоантигены).
Быстродействующий α2-антиплазмин обладает также антиактиваторным действием, но он не идентичен другому антиактиватору, описанному Hedner (1973, 1977 гг.).
Из других ингибиторов фибринолиза, обладающих значительно более слабым действием, заслуживают упоминания α2-макроглобулин и ингибитор С1-эстеразы. Последний ингибирует фактор ХIIа, калликреин и отчасти плазмин, специфически блокирует внутренний (ХIIа-зависимый) фибринолиз. Вместе с тем имеются данные о том, что α2-макроглобулин не столько препятствует фибринолизу, сколько защищает плазмин от других, более мощных, ингибиторов. В частности, комплекс макроглобулин-плазмин защищен от быстродействующего α2-антиплазмина, благодаря чему при активации плазминовой системы идет лизис не только фибрина и РФМК, но в небольшой степени и фибриногена, хотя в плазме имеется избыток α2-антиплазмина.
Плазминовая система специфически адаптирована к лизису фибрина и растворимых фибрин-мономерных комплексов (РФМК), хотя при ее значительной активации расщеплению подвергаются и другие белки (в том числе факторы свертывания V и VIII).
Механизм преимущественной активации фибринолиза в тромбах и сгустках, резко выраженного преобладания фибринолиза над фибриногенолизом пока не может считаться окончательно выясненным. Твердо доказана лишь способность частично активированного плазминогена (лиз-плазминогена) связываться с фибрином. Установлено также, что растворение идет тем быстрее, чем выше локальная концентрация в сгустках плазминогена. Особенно важно, что сосудистый активатор плазминогена также концентрируется на фибрине. Наконец, установлено, что α2-антиплазмин намного слабее инактивирует связанный с фибрином плазмин, тогда как циркулирующий «свободный» плазмин образует с этим мощным ингибитором плохо диссоциирующие комплексы.
Схема 3 Фибринолитическая система
1) нативный плазминоген с NH2-терминальной глютаминовой кислотой – «глю-плазминоген»;
2) подвергшийся ограниченному протеолизу плазминоген с NH2-терминальным лизином, валином или метионином – «лиз-плазминоген». Лиз-плазминоген в 10–20 раз быстрее трансформируется активаторами в плазмин, имеет значительно более выраженное, чем глю-плазминоген, сродство к фибрину и значительно быстрее последнего метаболизируется – его Т1/2 в циркуляции около 0,8 суток, а глю-плазминогена – 1,24 ± 0,29 суток. По механизму протеолитического действия плазмин наиболее близок к трипсину.
После активации плазминоген быстро исчезает из кровотока – блокируется антиплазминами и удаляется. Вслед за введением больших доз стрептокиназы или урокиназы уровень плазминогена в крови снижается до нуля, но затем в течение 12–28 ч восстанавливается, если прекращена его дальнейшая активация.
Эта способность активаторов фибринолиза быстро истощать запасы плазминогена в крови и на время оставлять больного без ферментативного фибринолиза важна для клиники и должна учитываться при лечении тромбозов и синдрома диссеминированного внутрисосудистого свертывания крови.
Существующие в организме механизмы активации плазминогена весьма разнообразны, но, подобно механизмам свертывания крови, они также могут быть подразделены на две основные группы – с внутренней и внешней активацией.
Ведущий внутренний механизм запускается теми же факторами, какие инициируют свертывание крови, а именно фактором ХIIа, который, взаимодействуя с прекалликреином и высокомолекулярным кининогеном плазмы (ВМК), активирует плазминоген. Такой путь фибринолиза – базисный, обеспечивающий активацию плазминовой системы не вслед за свертыванием крови, а одновременно с ним. Он функционирует по «замкнутому циклу», поскольку образующиеся первые порции калликреина и плазмина вызывают протеолиз фактора XII, отцепляя фрагменты, под действием которых нарастает изменение прекалликреина в калликреин. Такая интенсивная самоактивация приводит к тому, что ХIIа-калликреин-зависимый фибринолиз при интенсивном внутрисосудистом свертывании крови истощается быстро, раньше других механизмов фибринолиза.
Лимитирующими факторами являются в первую очередь ВМК и прекалликреин. Их плазменный резерв быстро истощается, тогда как уровень плазминогена остается в крови еще достаточно высоким. В таких условиях ХIIа-зависимый фибринолиз уже не функционирует, но поддается другим (не калликреиновым) способам активации – стрептокиназой и урокиназой. Лишь вслед за этим возможно истощение запасов плазминогена, что делает неэффективным любые способы активации плазминовой системы. Определенное участие в активации внутреннего механизма фибринолиза принимает, по-видимому, и фактор Виллебранда. В частности, на образцах плазмы с дефицитом ВМК показано, что фактор Виллебранда в 2–3 раза усиливает превращение прекалликреина в калликреин под влиянием фрагментов фактора XII. В присутствии ВМК, обладающего значительно более мощным влиянием на активацию прекалликреина, это действие фактора Виллебранда становится малоощутимым.
Заслуживает внимания то обстоятельство, что если в свертывании крови компонентам калликреин-кининовой системы отводится в определенной мере вспомогательная функция, то в гуморальном механизме фибринолиза это один из ведущих механизмов. Возможно, именно поэтому при генетически обусловленном дефиците плазменного прекалликреина (дефект Флетчера) или ВМК (дефект Фитцжеральда – Вильсона) у больных нет кровоточивости и вместе с тем прослеживается наклонность к тромбозам.
Важнейшими стимуляторами внешнего механизма фибринолиза являются белковые активаторы плазминогена, синтезируемые в сосудистой стенке. Эти активаторы подразделяются на высокомолекулярные и низкомолекулярные фракции, обнаруживают высокое сродство к фибрину. Физиологическая регуляция синтеза и выделения в кровь сосудистых активаторов изучена недостаточно. Тем не менее известно, что их интенсивный выброс происходит при нарушении проходимости сосудов, в том числе и при пережатии сосуда манжетой, а также при физических нагрузках, под влиянием вазоактивных веществ. Определение эуглобулинового лизиса до и после пережатия сосуда (манжеточная проба) используется для оценки резерва сосудистых активаторов плазминогена и функциональной полноценности механизмов их либерации. Депрессия данных механизмов характерна для ряда тромбофилических состояний. Стероидные гормоны анаболического действия повышают синтез в эндотелии активаторов фибринолиза, с чем отчасти связывается их благоприятное влияние на течение флеботромботической болезни.
Мощные активаторы плазминогена содержатся также в клетках крови – эритроцитах, тромбоцитах и особенно лейкоцитах. При внутрисосудистом свертывании крови, тромбообразовании, воздействии эндотоксином, активации системы комплемента, гемолизе эти активаторы освобождаются из клеток в «плазматическую атмосферу» и активируют плазминоген.
Более того, установлено, что гранулоциты секретируют не только активатор плазминогена, но и внутриклеточные протеазы (цитокиназы), которые самостоятельно, без участия плазмина, переваривают фибрин. При этом образуются иные продукты расщепления фибрина, чем при его плазминовом расщеплении.
Следовательно, лейкоциты обеспечивают функционирование самостоятельного (неплазминового) механизма растворения фибрина. Этот альтернативный механизм играет важную роль в ограничении размеров тромбов и в деблокировании микроциркуляторного русла при диссеминированном внутрисосудистом свертывании крови.
Разнообразные активаторы плазминогена (цитокиназы) содержатся и в других тканях и клетках, особенно в эпителиальной, мышечной и мезенхимальной, а также в секретах и экскретах – моче, молоке, желчи, слюне. Некоторые из них поступают в определенных количествах в кровь, участвуя в активации плазминогена. В частности, таким свойством обладает урокиназа – активатор фибринолиза, синтезируемый в почечном эпителии и выделяющийся с мочой. В кровь поступает небольшое количество урокиназы, ответственное приблизительно за 10–15% общей плазминоген-активаторной функции. В настоящее время установлено, что большинство тканевых активаторов плазминоген идентично сосудистому, эндотелиальному.
Фибринолиз ингибируется рядом антиактиваторов и антиплазминов, из которых наиболее важен недавно открытый быстродействующий антиплазмин, относящийся к α2-глобулинам (молекулярная масса 65 000–70 000) и содержащийся в плазме в количестве 70 мг/л. Этого количества достаточно, чтобы нейтрализовать более 2/3 всего плазмина, образующегося при максимальной активации плазминогена. Однако плазмин, связанный с фибрином, хуже комплексируется с антиплазмином, чем при циркуляции в свободном состоянии. Антиплазмин ослабляет процесс связывания плазминогена с фибрином. Присутствие в плазме циркулирующих комплексов плазмин – антиплазмин, как и комплексов тромбин-ATIII, служит признаком интенсивного внутрисосудистого свертывания крови и активации фибринолиза. Выявление этих комплексов облегчается тем, что в них появляются новые антигенные свойства (так называемые неоантигены).
Быстродействующий α2-антиплазмин обладает также антиактиваторным действием, но он не идентичен другому антиактиватору, описанному Hedner (1973, 1977 гг.).
Из других ингибиторов фибринолиза, обладающих значительно более слабым действием, заслуживают упоминания α2-макроглобулин и ингибитор С1-эстеразы. Последний ингибирует фактор ХIIа, калликреин и отчасти плазмин, специфически блокирует внутренний (ХIIа-зависимый) фибринолиз. Вместе с тем имеются данные о том, что α2-макроглобулин не столько препятствует фибринолизу, сколько защищает плазмин от других, более мощных, ингибиторов. В частности, комплекс макроглобулин-плазмин защищен от быстродействующего α2-антиплазмина, благодаря чему при активации плазминовой системы идет лизис не только фибрина и РФМК, но в небольшой степени и фибриногена, хотя в плазме имеется избыток α2-антиплазмина.
Плазминовая система специфически адаптирована к лизису фибрина и растворимых фибрин-мономерных комплексов (РФМК), хотя при ее значительной активации расщеплению подвергаются и другие белки (в том числе факторы свертывания V и VIII).
Механизм преимущественной активации фибринолиза в тромбах и сгустках, резко выраженного преобладания фибринолиза над фибриногенолизом пока не может считаться окончательно выясненным. Твердо доказана лишь способность частично активированного плазминогена (лиз-плазминогена) связываться с фибрином. Установлено также, что растворение идет тем быстрее, чем выше локальная концентрация в сгустках плазминогена. Особенно важно, что сосудистый активатор плазминогена также концентрируется на фибрине. Наконец, установлено, что α2-антиплазмин намного слабее инактивирует связанный с фибрином плазмин, тогда как циркулирующий «свободный» плазмин образует с этим мощным ингибитором плохо диссоциирующие комплексы.
Схема 3 Фибринолитическая система
Часть II. Частная гематология
Глава 1. Анемии
Анемия – уменьшение общего количества гемоглобина, чаще всего проявляющееся уменьшением его концентрации в единице объема крови. Все острые кровопотери обязательно сопровождаются признаками анемии: бледностью, одышкой, сердцебиением при небольших нагрузках, хотя при этом еще не наступает разбавления крови плазмой, вызывающего падение концентрации гемоглобина. Нередко наблюдается и обратная ситуация. Например, при беременности (или сердечной недостаточности) количество жидкой части крови возрастает (гидремия), падает концентрация гемоглобина и эритроцитов, без общего уменьшения массы гемоглобина и эритроцитов.
В большинстве случаев, за исключением железодефицитных состояний и талассемии, анемия сопровождается и снижением содержания красных кровяных телец в 1 л крови.
Поскольку анемия является частным симптомом какого-то общего заболевания, строгая классификация анемий невозможна. Врачу удобно делить анемии на гипо– и гиперхромные, так как цветовой показатель позволяет направить диагностический поиск в нужном направлении. Однако классическая гиперхромная витамин-В12-дефицитная анемия нередко бывает нормохромной, а при гипохромной железодефицитной анемии субъективные симптомы появляются до того, как снизится цветовой показатель. Классифицирование анемии на основе ее механизма развития, как и по причине, ее вызвавшей (нозологическое классифицирование), также не может быть выдержано до конца. Например, постгеморрагическая железодефицитная анемия, представляющая собой одно из самых массовых заболеваний человечества, может быть обусловлена меноррагиями (потерей крови в период менструации у женщин), не компенсируемыми железом пищи, хроническими кровопотерями при геморрое или при носовых кровотечениях.
Постгеморрагическая железодефицитная анемия отнесена к группе анемий, связанных с кровопотерей, и к группе анемий, связанных с нарушенным кровообразованием.
В большинстве случаев, за исключением железодефицитных состояний и талассемии, анемия сопровождается и снижением содержания красных кровяных телец в 1 л крови.
Поскольку анемия является частным симптомом какого-то общего заболевания, строгая классификация анемий невозможна. Врачу удобно делить анемии на гипо– и гиперхромные, так как цветовой показатель позволяет направить диагностический поиск в нужном направлении. Однако классическая гиперхромная витамин-В12-дефицитная анемия нередко бывает нормохромной, а при гипохромной железодефицитной анемии субъективные симптомы появляются до того, как снизится цветовой показатель. Классифицирование анемии на основе ее механизма развития, как и по причине, ее вызвавшей (нозологическое классифицирование), также не может быть выдержано до конца. Например, постгеморрагическая железодефицитная анемия, представляющая собой одно из самых массовых заболеваний человечества, может быть обусловлена меноррагиями (потерей крови в период менструации у женщин), не компенсируемыми железом пищи, хроническими кровопотерями при геморрое или при носовых кровотечениях.
Постгеморрагическая железодефицитная анемия отнесена к группе анемий, связанных с кровопотерей, и к группе анемий, связанных с нарушенным кровообразованием.
Острая постгеморрагическая анемия
Под острой постгеморрагической анемией понимают анемию, развившуюся в результате быстрой потери значительного количества крови.
В механизме развития основных симптомов острой кровопотери ведущую роль играет быстрое уменьшение общего объема крови, прежде всего ее плазмы. Уменьшение объема эритроцитов ведет к острой гипоксии, которая клинически проявляется появлением одышки, сердцебиением.
Коллапс (обморочное состояние) или гипотония (снижение артериального давления) вызваны в основном потерей плазмы. Во время кровотечения и сразу после него отмечаются выброс надпочечниками катехоламинов, что вызывает спазм периферических сосудов. Уменьшение объема сосудистого русла способствует компенсации снижения объема циркулирующей крови. Однако длительный спазм периферических сосудов неблагоприятно действует на микроциркуляцию и может привести к развитию шока. Один из главных механизмов саморегуляции организма – восстановление объема крови путем мобилизации собственной межтканевой жидкости и ее выброса в сосудистое русло. Данный процесс носит название аутогемодилюции. Если аутогемодилюция выражена недостаточно или истощается, то наступает декомпенсация, и без лечения больной погибает. В результате гипоксии, связанной с кровопотерей, повышается содержание эритропоэтина, следствием чего становятся повышенное образование чувствительных к нему клеток и выброс ретикулоцитов.
Клиника
Острая постгеморрагическая анемия вызывает прежде всего симптомы коллапса. У больного наблюдаются резкая слабость, головокружение, бледность, сухость во рту, холодный пот, рвота. Снижается артериальное и венозное давление, уменьшается сердечный выброс крови, резко учащается пульс. Наполнение пульса становится слабым.
Клиническая картина определяется количеством потерянной крови, скоростью ее истечения и в какой-то мере зависит и от источника кровопотери. Имеются данные о неодинаковой компенсации в зависимости от источника кровотечения.
Для оценки кровопотери рекомендуется использовать формулу:
К – коэффициент, равный 27 при желудочно-кишечной кровопотере, 33 – при полостных кровотечениях, 24 – при ранениях конечностей и 22 – при поражении грудной клетки;
ШИ – шоковый индекс, равный отношению частоты пульса к систолическому давлению.
В первые часы при большой кровопотере может быть незначительное снижение уровня гемоглобина и эритроцитов, соответственно не уменьшен гематокрит (часть объема крови, приходящаяся на форменные элементы), и лишь исследование объема циркулирующих эритроцитов может выявить его значительное снижение.
Если кровотечение удалось остановить, то через 2–3 дня наблюдается снижение уровня гемоглобина и эритроцитов вследствие проникновения в кровь тканевой жидкости, поэтому в первое время после кровопотери малокровие имеет нормохромный характер. Содержание тромбоцитов в период кровотечения может быть сниженным в связи с их потреблением в процессе тромбообразования.
В основе диагностики скрытого массивного кровотечения лежат клинические проявления, подкрепленные некоторыми лабораторными данными (пробами Грегерсена, Вебера, повышением уровня остаточного азота в случае кровотечения из верхних отделов пищеварительного тракта).
Лечение
Лечение острой постгеморрагической анемии начинается с остановки кровотечения и проведения противошоковых мероприятий. Показаниями к переливанию крови при острой кровопотере являются: продолжительное кровотечение, значительное падение цифр систолического артериального давления до 90 мм рт. ст. и ниже, учащение пульса по сравнению с нормой на 20 ударов в минуту и более. Кровопотеря в пределах 10–15% исходного объема циркулирующей крови (ОЦК) не требует кровевозмещения, а потеря даже 25% ОЦК требует лишь небольшой коррекции. Переливание кровезаменителей проводится больным с потерей более 25% крови. Для заместительной терапии используют полиглюкин в объеме до 2 л/сут. С целью улучшения микроциркуляции используют внутривенное введение реополиглюкина, желатиноля или альбуминов. Эритроцитную массу в объеме 30–40% кровопотери следует использовать только после восстановления кровообращения посредством восполнения ОЦК указанными выше растворами. Для улучшения реологических свойств крови эритроцитную массу разводят реополиглюкином или 5%-ным раствором альбумина в соотношении 1 : 1.
При массивной кровопотере большое значение имеет скорость переливания. Обычно венозное давление резко снижено, локтевые вены спавшиеся, поэтому следует прибегать к пункции подключичных вен или веносекциям с последующим струйным введением растворов в 2–3 вены. Следует подчеркнуть недопустимость восполнения всей кровопотери кровью во избежание «синдрома массивных трансфузий». Необходимо помнить также о коррекции белков плазмы, для чего используют альбумин или протеин. С целью коррекции водного баланса организма производят внутривенные вливания 0,9%-ного раствора хлорида натрия, 5%-ного раствора глюкозы, раствора Рингера – Локка. Для нормализации рН крови используется лактасол.
Переливание цельной крови, как правило, нецелесообразно.
В механизме развития основных симптомов острой кровопотери ведущую роль играет быстрое уменьшение общего объема крови, прежде всего ее плазмы. Уменьшение объема эритроцитов ведет к острой гипоксии, которая клинически проявляется появлением одышки, сердцебиением.
Коллапс (обморочное состояние) или гипотония (снижение артериального давления) вызваны в основном потерей плазмы. Во время кровотечения и сразу после него отмечаются выброс надпочечниками катехоламинов, что вызывает спазм периферических сосудов. Уменьшение объема сосудистого русла способствует компенсации снижения объема циркулирующей крови. Однако длительный спазм периферических сосудов неблагоприятно действует на микроциркуляцию и может привести к развитию шока. Один из главных механизмов саморегуляции организма – восстановление объема крови путем мобилизации собственной межтканевой жидкости и ее выброса в сосудистое русло. Данный процесс носит название аутогемодилюции. Если аутогемодилюция выражена недостаточно или истощается, то наступает декомпенсация, и без лечения больной погибает. В результате гипоксии, связанной с кровопотерей, повышается содержание эритропоэтина, следствием чего становятся повышенное образование чувствительных к нему клеток и выброс ретикулоцитов.
Клиника
Острая постгеморрагическая анемия вызывает прежде всего симптомы коллапса. У больного наблюдаются резкая слабость, головокружение, бледность, сухость во рту, холодный пот, рвота. Снижается артериальное и венозное давление, уменьшается сердечный выброс крови, резко учащается пульс. Наполнение пульса становится слабым.
Клиническая картина определяется количеством потерянной крови, скоростью ее истечения и в какой-то мере зависит и от источника кровопотери. Имеются данные о неодинаковой компенсации в зависимости от источника кровотечения.
Для оценки кровопотери рекомендуется использовать формулу:
П = K + 44lgШИ,Где П – потеря крови в процентах;
К – коэффициент, равный 27 при желудочно-кишечной кровопотере, 33 – при полостных кровотечениях, 24 – при ранениях конечностей и 22 – при поражении грудной клетки;
ШИ – шоковый индекс, равный отношению частоты пульса к систолическому давлению.
В первые часы при большой кровопотере может быть незначительное снижение уровня гемоглобина и эритроцитов, соответственно не уменьшен гематокрит (часть объема крови, приходящаяся на форменные элементы), и лишь исследование объема циркулирующих эритроцитов может выявить его значительное снижение.
Если кровотечение удалось остановить, то через 2–3 дня наблюдается снижение уровня гемоглобина и эритроцитов вследствие проникновения в кровь тканевой жидкости, поэтому в первое время после кровопотери малокровие имеет нормохромный характер. Содержание тромбоцитов в период кровотечения может быть сниженным в связи с их потреблением в процессе тромбообразования.
В основе диагностики скрытого массивного кровотечения лежат клинические проявления, подкрепленные некоторыми лабораторными данными (пробами Грегерсена, Вебера, повышением уровня остаточного азота в случае кровотечения из верхних отделов пищеварительного тракта).
Лечение
Лечение острой постгеморрагической анемии начинается с остановки кровотечения и проведения противошоковых мероприятий. Показаниями к переливанию крови при острой кровопотере являются: продолжительное кровотечение, значительное падение цифр систолического артериального давления до 90 мм рт. ст. и ниже, учащение пульса по сравнению с нормой на 20 ударов в минуту и более. Кровопотеря в пределах 10–15% исходного объема циркулирующей крови (ОЦК) не требует кровевозмещения, а потеря даже 25% ОЦК требует лишь небольшой коррекции. Переливание кровезаменителей проводится больным с потерей более 25% крови. Для заместительной терапии используют полиглюкин в объеме до 2 л/сут. С целью улучшения микроциркуляции используют внутривенное введение реополиглюкина, желатиноля или альбуминов. Эритроцитную массу в объеме 30–40% кровопотери следует использовать только после восстановления кровообращения посредством восполнения ОЦК указанными выше растворами. Для улучшения реологических свойств крови эритроцитную массу разводят реополиглюкином или 5%-ным раствором альбумина в соотношении 1 : 1.
При массивной кровопотере большое значение имеет скорость переливания. Обычно венозное давление резко снижено, локтевые вены спавшиеся, поэтому следует прибегать к пункции подключичных вен или веносекциям с последующим струйным введением растворов в 2–3 вены. Следует подчеркнуть недопустимость восполнения всей кровопотери кровью во избежание «синдрома массивных трансфузий». Необходимо помнить также о коррекции белков плазмы, для чего используют альбумин или протеин. С целью коррекции водного баланса организма производят внутривенные вливания 0,9%-ного раствора хлорида натрия, 5%-ного раствора глюкозы, раствора Рингера – Локка. Для нормализации рН крови используется лактасол.
Переливание цельной крови, как правило, нецелесообразно.
Железодефицитные анемии
Железодефицитные анемии – широко распространенные болезни, при которых снижается содержание железа в сыворотке крови, в костном мозге, а также в его депо. Вследствие этого нарушается продуцирование гемоглобина, а в дальнейшем – и эритроцитов, появляются гипохромная анемия и трофические нарушения в тканях.
Причины и механизм развития заболевания
Наиболее частой причиной железодефицитной анемии являются кровопотери, особенно длительные, постоянные, хотя и незначительные. Организм теряет больше железа, чем получает из пищи.
Физиологическое всасывание железа из пищи ограничено. Обычно мужчины получают с пищей 18 мг железа, из которых может всасываться 1–1,5 мг, женщины – 12–15 мг железа, из которых всасывается 1–1,3 мг. При повышенных потребностях организма в железе из пищи может всосаться максимум 2–2,5 мг. Следовательно, дефицит железа развивается тогда, когда организм теряет его более 2 мг/сут. Физиологические потери железа у мужчин с мочой, калом, потом, слущивающимся эпителием кожи не превышают 1 мг, поэтому при нормальном количестве железа в пище, правильном питании и нормальном кишечном всасывании и без кровопотерь у мужчин не должна появиться недостаточность железа. У женщин к тем расходам, что и у мужчин, прибавляются потери железа с кровью во время менструаций, беременности, родов, лактации. В связи с этим у женщин очень часто потребности в железе превышают всасывание железа из пищи. Это и становится наиболее частой причиной железодефицитных анемий.
Причины и механизм развития заболевания
Наиболее частой причиной железодефицитной анемии являются кровопотери, особенно длительные, постоянные, хотя и незначительные. Организм теряет больше железа, чем получает из пищи.
Физиологическое всасывание железа из пищи ограничено. Обычно мужчины получают с пищей 18 мг железа, из которых может всасываться 1–1,5 мг, женщины – 12–15 мг железа, из которых всасывается 1–1,3 мг. При повышенных потребностях организма в железе из пищи может всосаться максимум 2–2,5 мг. Следовательно, дефицит железа развивается тогда, когда организм теряет его более 2 мг/сут. Физиологические потери железа у мужчин с мочой, калом, потом, слущивающимся эпителием кожи не превышают 1 мг, поэтому при нормальном количестве железа в пище, правильном питании и нормальном кишечном всасывании и без кровопотерь у мужчин не должна появиться недостаточность железа. У женщин к тем расходам, что и у мужчин, прибавляются потери железа с кровью во время менструаций, беременности, родов, лактации. В связи с этим у женщин очень часто потребности в железе превышают всасывание железа из пищи. Это и становится наиболее частой причиной железодефицитных анемий.