гипотеза Праута.
Однако таблица Берцелиуса, казалось, разрушила это привлекательное предположение (привлекательное потому, что, подобно античным ученым, Праут сводил все возраставшее число элементов к одному основному веществу и, таким образом, как будто бы придавал Вселенной упорядоченность и симметрию). Однако, если принять атомный вес водорода («основы»), равным 1, то атомный вес кислорода составит приблизительно 15.9 веса водорода, но едва ли можно согласиться с тем, что кислород состоит из 15 плюс еще 9/10 атома водорода.
В следующем столетии таблицы атомных весов постоянно уточнялись, и все более очевидными становились выводы Берцелиуса, считавшего, что атомные веса различных элементов не являются целыми числами, кратными атомному весу водорода.
В шестидесятых годах XIX в. бельгийский химик Жан Сервэ Стас (1813—1891) определил атомные веса точнее, чем Берцелиус. В начале XX в. американский химик Теодор Уильям Ричардс (1868—1928), приняв все меры предосторожности (во многом надуманные), определил величины атомных весов с такой точностью, которая только возможна при использовании чисто химических методов. Исследования Стаса и Ричардса ответили на те вопросы, которые в работах Берцелиуса оставались нерешенными.
Нельзя было не принять тот факт, что атомные веса выражаются нецелыми числами, и в свете этого гипотеза Праута, казалась бы, все более теряла смысл. Однако в то время, когда Ричарде проводил свои поразительно точные определения атомных весов, вновь встал вопрос о том, что следует понимать под атомным весом. И на этом этапе развития химии гипотезе Праута, как мы увидим далее, суждено было возродиться.
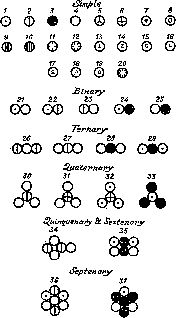
Рис. 9. Символы некоторых элементов и соединений, предложенные Дальтоном; 1— водород; 3— углерод; 4— кислород; 15— медь; 17— серебро; 19— золото; 21— вода. Дальтон дал неверную формулу воды (НО вместо Н 2О), но его формулы монооксида ( 25) и диоксида углерода ( 28) верны.
Поскольку, как выяснилось, атомные веса различных элементов взаимосвязаны не столь простым образом, как это ранее предполагалось, необходимо было выявить стандарт, исходя из которого можно было бы определять атомные веса элементов. Естественным казалось принять за единицу атомного веса атомный вес водорода, как это сделали Берцелиус и Дальтон. Но при этом атомный вес кислорода выражался неудобным нецелым числом 15.9, а ведь именно кислород обычно использовался для определения соотношений элементов в различных соединениях.
Чтобы атомный вес кислорода выражался удобным целым числом при минимальном нарушении стандарта, т. е. атомного веса водорода, атомный вес кислорода округлили и приняли равным 16.000 (вместо 15.9). Таким образом, в качестве стандарта был принят атомный вес кислорода, равный 16; атомный вес водорода при этом оказался равным 1.008. Атомный вес кислорода служил стандартом вплоть до середины XX в.
После того как атомистическая теория была принята, стало возможным изображать вещества в виде молекул, содержащих постоянное число атомов различных элементов. Вполне естественным было попытаться изобразить такие молекулы в виде набора маленьких кружков, представляющих собой атомы; при этом атомы каждого вида можно было изобразить кружками определенного типа.
Дальтон пытался ввести именно эту символику. Простым кружком он изображал атом кислорода; кружком с точкой посередине — атом водорода; кружком с вертикальной линией — атом азота; закрашенным черным кружком — атом углерода и т. д. Поскольку придумывать различные типы кружков становилось все труднее и труднее, Дальтон стал использовать начальные буквы названий элементов. Так, серу он изображал в виде кружка с буквой S, фосфор — в виде кружка с буквой P и т. д.
Берцелиус решил, что кружки излишни, достаточно лишь начальных букв. Он предложил, чтобы каждому элементу соответствовал свой особый знак, который был бы одновременно и символом элемента, и символом одиночного атома этого элемента, и в качестве такого знака предложил использовать начальную букву латинского названия элемента. (К счастью, для англоязычных народов латинское название почти всегда похоже на английское.) В тех случаях, когда названия двух или более элементов начинались с одних и тех же начальных букв, добавлялась вторая буква названия. Так появились химические символыэлементов, которыми пользуются во всем мире и поныне.
Итак, химическим символом углерода, водорода, кислорода, азота, фосфора и серы стали соответственно C, H, O, N, P и S, кальций и хлор (углерод первым завладел прописной буквой C) обозначались соответственно Ca и Cl.
С помощью химических символов легко показать количество атомов в молекуле. Так, молекулу водорода, состоящую из двух атомов водорода, записывают как H 2, а молекулу воды, содержащую два атома водорода и один атом кислорода,— как H 2O. (Знак без числового индекса, это легко увидеть, означает единичный атом.) Углекислый газ — это CO 2, серная кислота — H 2SO 4, а хлорид водорода — HCl. Химические формулыэтих простых соединений говорят сами за себя.
Химические формулы можно объединять в химические уравнения, описывающие реакции. С помощью такого уравнения можно, например, показать, что углерод соединяется с кислородом и образует углекислый газ:
C + O 2» CO 2.
В таких уравнениях, чтобы не нарушить закона сохранения массы веществ, необходимо учитывать все участвующие в реакции атомы.
Предположим, мы хотим сказать, что водород соединяется с хлором и образует хлорид водорода. Если это записать просто как
H 2+ Cl 2» HCl,
то нетрудно заметить, что среди исходных веществ у нас два атома водорода и два атома хлора, а среди продуктов реакции — только по одному. Чтобы уравнять правую и левую части, перед формулами исходных веществ и продуктов реакции ставят коэффициенты. В результате реакция образования хлорида водорода записывается как
H 2+ Cl 2» 2HCl,
а реакция образования воды — как
2H 2+ O 2» 2H 2O.
Электролиз
Глава 6 Органическая химия
«Кирпичики» жизни
Однако таблица Берцелиуса, казалось, разрушила это привлекательное предположение (привлекательное потому, что, подобно античным ученым, Праут сводил все возраставшее число элементов к одному основному веществу и, таким образом, как будто бы придавал Вселенной упорядоченность и симметрию). Однако, если принять атомный вес водорода («основы»), равным 1, то атомный вес кислорода составит приблизительно 15.9 веса водорода, но едва ли можно согласиться с тем, что кислород состоит из 15 плюс еще 9/10 атома водорода.
В следующем столетии таблицы атомных весов постоянно уточнялись, и все более очевидными становились выводы Берцелиуса, считавшего, что атомные веса различных элементов не являются целыми числами, кратными атомному весу водорода.
В шестидесятых годах XIX в. бельгийский химик Жан Сервэ Стас (1813—1891) определил атомные веса точнее, чем Берцелиус. В начале XX в. американский химик Теодор Уильям Ричардс (1868—1928), приняв все меры предосторожности (во многом надуманные), определил величины атомных весов с такой точностью, которая только возможна при использовании чисто химических методов. Исследования Стаса и Ричардса ответили на те вопросы, которые в работах Берцелиуса оставались нерешенными.
Нельзя было не принять тот факт, что атомные веса выражаются нецелыми числами, и в свете этого гипотеза Праута, казалась бы, все более теряла смысл. Однако в то время, когда Ричарде проводил свои поразительно точные определения атомных весов, вновь встал вопрос о том, что следует понимать под атомным весом. И на этом этапе развития химии гипотезе Праута, как мы увидим далее, суждено было возродиться.
Рис. 9. Символы некоторых элементов и соединений, предложенные Дальтоном; 1— водород; 3— углерод; 4— кислород; 15— медь; 17— серебро; 19— золото; 21— вода. Дальтон дал неверную формулу воды (НО вместо Н 2О), но его формулы монооксида ( 25) и диоксида углерода ( 28) верны.
Поскольку, как выяснилось, атомные веса различных элементов взаимосвязаны не столь простым образом, как это ранее предполагалось, необходимо было выявить стандарт, исходя из которого можно было бы определять атомные веса элементов. Естественным казалось принять за единицу атомного веса атомный вес водорода, как это сделали Берцелиус и Дальтон. Но при этом атомный вес кислорода выражался неудобным нецелым числом 15.9, а ведь именно кислород обычно использовался для определения соотношений элементов в различных соединениях.
Чтобы атомный вес кислорода выражался удобным целым числом при минимальном нарушении стандарта, т. е. атомного веса водорода, атомный вес кислорода округлили и приняли равным 16.000 (вместо 15.9). Таким образом, в качестве стандарта был принят атомный вес кислорода, равный 16; атомный вес водорода при этом оказался равным 1.008. Атомный вес кислорода служил стандартом вплоть до середины XX в.
После того как атомистическая теория была принята, стало возможным изображать вещества в виде молекул, содержащих постоянное число атомов различных элементов. Вполне естественным было попытаться изобразить такие молекулы в виде набора маленьких кружков, представляющих собой атомы; при этом атомы каждого вида можно было изобразить кружками определенного типа.
Дальтон пытался ввести именно эту символику. Простым кружком он изображал атом кислорода; кружком с точкой посередине — атом водорода; кружком с вертикальной линией — атом азота; закрашенным черным кружком — атом углерода и т. д. Поскольку придумывать различные типы кружков становилось все труднее и труднее, Дальтон стал использовать начальные буквы названий элементов. Так, серу он изображал в виде кружка с буквой S, фосфор — в виде кружка с буквой P и т. д.
Берцелиус решил, что кружки излишни, достаточно лишь начальных букв. Он предложил, чтобы каждому элементу соответствовал свой особый знак, который был бы одновременно и символом элемента, и символом одиночного атома этого элемента, и в качестве такого знака предложил использовать начальную букву латинского названия элемента. (К счастью, для англоязычных народов латинское название почти всегда похоже на английское.) В тех случаях, когда названия двух или более элементов начинались с одних и тех же начальных букв, добавлялась вторая буква названия. Так появились химические символыэлементов, которыми пользуются во всем мире и поныне.
Итак, химическим символом углерода, водорода, кислорода, азота, фосфора и серы стали соответственно C, H, O, N, P и S, кальций и хлор (углерод первым завладел прописной буквой C) обозначались соответственно Ca и Cl.
С помощью химических символов легко показать количество атомов в молекуле. Так, молекулу водорода, состоящую из двух атомов водорода, записывают как H 2, а молекулу воды, содержащую два атома водорода и один атом кислорода,— как H 2O. (Знак без числового индекса, это легко увидеть, означает единичный атом.) Углекислый газ — это CO 2, серная кислота — H 2SO 4, а хлорид водорода — HCl. Химические формулыэтих простых соединений говорят сами за себя.
Химические формулы можно объединять в химические уравнения, описывающие реакции. С помощью такого уравнения можно, например, показать, что углерод соединяется с кислородом и образует углекислый газ:
C + O 2» CO 2.
В таких уравнениях, чтобы не нарушить закона сохранения массы веществ, необходимо учитывать все участвующие в реакции атомы.
Предположим, мы хотим сказать, что водород соединяется с хлором и образует хлорид водорода. Если это записать просто как
H 2+ Cl 2» HCl,
то нетрудно заметить, что среди исходных веществ у нас два атома водорода и два атома хлора, а среди продуктов реакции — только по одному. Чтобы уравнять правую и левую части, перед формулами исходных веществ и продуктов реакции ставят коэффициенты. В результате реакция образования хлорида водорода записывается как
H 2+ Cl 2» 2HCl,
а реакция образования воды — как
2H 2+ O 2» 2H 2O.
Электролиз
Изучая влияние электрического тока на химические вещества, ученые смогли выделить ряд новых элементов. Вообще за полтора века, прошедшие с того времени, когда Бойль ввел понятие «элемент» (см. гл. 3), было открыто поразительно много веществ, отвечающих этому определению. Более того, было установлено, что некоторые простые и сложные вещества содержат неоткрытые элементы, которые химики не могли пока ни выделить, ни изучить.
Очень часто эти элементы входили в состав оксидов, т. е. соединений кислорода. Чтобы выделить элемент, соединенный с кислородом, последний необходимо было удалить. В принципе под воздействием какого-либо другого элемента, обладающего более сильным сродством к кислороду, атом (или атомы) кислорода может покинуть первый элемент и присоединиться ко второму. Этот метод оказался эффективным. Причем часто роль второго, отнимающего кислород, элемента выполнял углерод. Например, если железную руду, которая по сути является оксидом железа, нагревать на коксе (относительно чистая разновидность углерода), то углерод соединяется с кислородом; при этом образуются оксиды углерода и металлическое железо.
Рассмотрим теперь известь. По своим свойствам она тоже похожа на оксид. Однако ни один из известных тогда элементов, вступая в реакцию с кислородом, не образует известь. Следовательно, известь является оксидом неизвестного элемента. Пытаясь выделить этот неизвестный элемент, известь нагревали на коксе, но при этом ничего не происходило. Неизвестный элемент, по-видимому, так крепко удерживал кислород, что атомы углерода не могли оторвать от него атомы кислорода. Ни одно другое химическое вещество также не могло «заставить» известь отдать кислород.
Однако английский химик Гемфри Дэви (1778—1829) решил, что если вещество нельзя разложить химическим путем, то, возможно, это удастся осуществить под воздействием электрического тока: ведь таким способом удалось разложить даже молекулу воды.
Дэви сконструировал электрическую батарею, в которой насчитывалось более 250 металлических пластин; это была самая сильная из имевшихся в то время батарей. Пропуская ток, который давала эта батарея, через растворы соединений, предположительно содержащих неизвестные элементы, Дэви пытался таким образом выделить эти элементы, однако успеха не добился. Он только разложил воду и получил водород и кислород.
Очевидно, необходимо было прежде удалить воду. Однако через твердые вещества ему даже не удалось пропустить ток. Наконец, Дэви догадался расплавить соединения и пропустить ток через расплав.
Это оказалось действенным. 6 октября 1807 г. Дэви пропустил ток через расплавленный поташ (карбонат калия) и получил маленькие шарики металла, который он назвал потассием(от английского — potash). Этот металл, впоследствии названный калием, оказался очень активным. Он вытеснял кислород из воды, освобождая водород, причем реакция эта шла чрезвычайно бурно. Неделю спустя Дэви выделил из соды (карбоната натрия) содий(от английского — soda), впоследствии названный натрием. По своей активности, как выяснилось, натрий лишь незначительно уступает калию.
В 1808 г., пользуясь модифицированным вариантом метода Берцелиуса, Дэви выделил несколько металлов из их оксидов: магнийиз магнезии, стронцийиз оксида стронция, барийиз оксида бария и кальцийиз извести («кальций» — от латинских названий извести — calx, calcis).
Дэви также показал, что зеленоватый газ, который открывший его Шееле (см. гл. 4) считал оксидом, в действительности является элементом. Дэви предложил назвать его хлорин(от греческого ?????? — желто-зеленый). Позднее Гей-Люссак сократил это название до хлора. Дэви доказал, что соляная кислота, будучи сильной кислотой, не содержит атома кислорода в своей молекуле, и, таким образом, опроверг предположение Лавуазье, который рассматривал кислород как необходимый компонент всех кислот (см. гл. 4.)
Работы Дэви по электролизу продолжил его помощник и ученик Майкл Фарадей (1791—1867) [44], который впоследствии стал знаменитым ученым. Ряд электрохимических терминов, введенных Фарадеем, используется и по сей день (рис. 10). Так, например, он назвал расщепление молекул под действием электрического тока электролизом. По предложению специалиста по античной филологии Уильяма Уэвелла (1794—1866) Фарадей назвал соединение или раствор, способный проводить электрический ток, электролитом; металлические стержни или пластины, помещенные в расплавленный металл или раствор,— электродами; электрод, несущий положительный заряд,— анодом; электрод, несущий отрицательный заряд,— катодом.
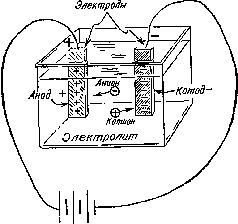
Рис. 10. Электролитический процесс Фарадей объяснял с помощью следующей схемы. Обозначения на рисунке соответствуют предложенной им терминологии.
Реально существующие частицы, благодаря которым электрический ток проходит через раствор или расплав, Фарадей назвал ионами(от греческого ??? — идущий). Ионы, перемещающиеся по направлению к аноду, он назвал анионами, а ионы, перемещающиеся по направлению к катоду,— катионами.
В 1832 г. Фарадей установил, что электрохимические процессы характеризуются определенными количественными соотношениями, и сформулировал следующие два закона электролиза. Вес вещества, выделившегося на электроде во время электролиза, пропорционален количеству электричества, пропущенного через раствор. Вес металла, выделенного данным количеством электричества, пропорционален эквивалентному весу этого металла.
Таким образом, если при взаимодействии серебра и калия с заданным количеством кислорода серебра в 2.7 раза больше, чем калия, то при данном количестве электричества серебра выделится в 2.7 раза больше, чем калия.
Законы Фарадея, по мнению некоторых химиков, указывали на то, что электричество, как и материю, можно разложить на постоянные минимальные единицы, или, другими словами, на «атомы электричества».
Предположим, что при пропускании электричества через раствор атомы материи притягиваются к катоду или к аноду «атомами электричества», и предположим, что для управления одним «атомом материи» во многих случаях достаточно одного «атома электричества», но иногда требуются два или даже три «атома электричества». Представив себе это, легко объяснить законы электролиза Фарадея.
Однако справедливость этого предположения была подтверждена только в самом конце XIX в., и тогда же было введено понятие «атомы электричества». Сам Фарадей никогда не проявлял энтузиазма по поводу «атомов электричества», да и атомистического учения в целом. [45]
Очень часто эти элементы входили в состав оксидов, т. е. соединений кислорода. Чтобы выделить элемент, соединенный с кислородом, последний необходимо было удалить. В принципе под воздействием какого-либо другого элемента, обладающего более сильным сродством к кислороду, атом (или атомы) кислорода может покинуть первый элемент и присоединиться ко второму. Этот метод оказался эффективным. Причем часто роль второго, отнимающего кислород, элемента выполнял углерод. Например, если железную руду, которая по сути является оксидом железа, нагревать на коксе (относительно чистая разновидность углерода), то углерод соединяется с кислородом; при этом образуются оксиды углерода и металлическое железо.
Рассмотрим теперь известь. По своим свойствам она тоже похожа на оксид. Однако ни один из известных тогда элементов, вступая в реакцию с кислородом, не образует известь. Следовательно, известь является оксидом неизвестного элемента. Пытаясь выделить этот неизвестный элемент, известь нагревали на коксе, но при этом ничего не происходило. Неизвестный элемент, по-видимому, так крепко удерживал кислород, что атомы углерода не могли оторвать от него атомы кислорода. Ни одно другое химическое вещество также не могло «заставить» известь отдать кислород.
Однако английский химик Гемфри Дэви (1778—1829) решил, что если вещество нельзя разложить химическим путем, то, возможно, это удастся осуществить под воздействием электрического тока: ведь таким способом удалось разложить даже молекулу воды.
Дэви сконструировал электрическую батарею, в которой насчитывалось более 250 металлических пластин; это была самая сильная из имевшихся в то время батарей. Пропуская ток, который давала эта батарея, через растворы соединений, предположительно содержащих неизвестные элементы, Дэви пытался таким образом выделить эти элементы, однако успеха не добился. Он только разложил воду и получил водород и кислород.
Очевидно, необходимо было прежде удалить воду. Однако через твердые вещества ему даже не удалось пропустить ток. Наконец, Дэви догадался расплавить соединения и пропустить ток через расплав.
Это оказалось действенным. 6 октября 1807 г. Дэви пропустил ток через расплавленный поташ (карбонат калия) и получил маленькие шарики металла, который он назвал потассием(от английского — potash). Этот металл, впоследствии названный калием, оказался очень активным. Он вытеснял кислород из воды, освобождая водород, причем реакция эта шла чрезвычайно бурно. Неделю спустя Дэви выделил из соды (карбоната натрия) содий(от английского — soda), впоследствии названный натрием. По своей активности, как выяснилось, натрий лишь незначительно уступает калию.
В 1808 г., пользуясь модифицированным вариантом метода Берцелиуса, Дэви выделил несколько металлов из их оксидов: магнийиз магнезии, стронцийиз оксида стронция, барийиз оксида бария и кальцийиз извести («кальций» — от латинских названий извести — calx, calcis).
Дэви также показал, что зеленоватый газ, который открывший его Шееле (см. гл. 4) считал оксидом, в действительности является элементом. Дэви предложил назвать его хлорин(от греческого ?????? — желто-зеленый). Позднее Гей-Люссак сократил это название до хлора. Дэви доказал, что соляная кислота, будучи сильной кислотой, не содержит атома кислорода в своей молекуле, и, таким образом, опроверг предположение Лавуазье, который рассматривал кислород как необходимый компонент всех кислот (см. гл. 4.)
Работы Дэви по электролизу продолжил его помощник и ученик Майкл Фарадей (1791—1867) [44], который впоследствии стал знаменитым ученым. Ряд электрохимических терминов, введенных Фарадеем, используется и по сей день (рис. 10). Так, например, он назвал расщепление молекул под действием электрического тока электролизом. По предложению специалиста по античной филологии Уильяма Уэвелла (1794—1866) Фарадей назвал соединение или раствор, способный проводить электрический ток, электролитом; металлические стержни или пластины, помещенные в расплавленный металл или раствор,— электродами; электрод, несущий положительный заряд,— анодом; электрод, несущий отрицательный заряд,— катодом.
Рис. 10. Электролитический процесс Фарадей объяснял с помощью следующей схемы. Обозначения на рисунке соответствуют предложенной им терминологии.
Реально существующие частицы, благодаря которым электрический ток проходит через раствор или расплав, Фарадей назвал ионами(от греческого ??? — идущий). Ионы, перемещающиеся по направлению к аноду, он назвал анионами, а ионы, перемещающиеся по направлению к катоду,— катионами.
В 1832 г. Фарадей установил, что электрохимические процессы характеризуются определенными количественными соотношениями, и сформулировал следующие два закона электролиза. Вес вещества, выделившегося на электроде во время электролиза, пропорционален количеству электричества, пропущенного через раствор. Вес металла, выделенного данным количеством электричества, пропорционален эквивалентному весу этого металла.
Таким образом, если при взаимодействии серебра и калия с заданным количеством кислорода серебра в 2.7 раза больше, чем калия, то при данном количестве электричества серебра выделится в 2.7 раза больше, чем калия.
Законы Фарадея, по мнению некоторых химиков, указывали на то, что электричество, как и материю, можно разложить на постоянные минимальные единицы, или, другими словами, на «атомы электричества».
Предположим, что при пропускании электричества через раствор атомы материи притягиваются к катоду или к аноду «атомами электричества», и предположим, что для управления одним «атомом материи» во многих случаях достаточно одного «атома электричества», но иногда требуются два или даже три «атома электричества». Представив себе это, легко объяснить законы электролиза Фарадея.
Однако справедливость этого предположения была подтверждена только в самом конце XIX в., и тогда же было введено понятие «атомы электричества». Сам Фарадей никогда не проявлял энтузиазма по поводу «атомов электричества», да и атомистического учения в целом. [45]
Глава 6 Органическая химия
Крушение витализма
Еще со времени открытия огня человек разделил вещества на две группы: горючие и негорючие. К горючим веществам относились, в частности, дерево и жир или масло, они в основном и служили топливом. Дерево — это продукт растительного происхождения, а жир и масло — продукты как животного, так и растительного происхождения. Вода, песок, различные горные породы и большинство других веществ минерального происхождения не горели, более того, гасили огонь.
Таким образом, между способностью вещества к горению и принадлежностью его к живому или неживому миру существовала определенная связь. Хотя, безусловно, были известны и исключения. Например, уголь и сера — продукты неживой материи — входили в группу горючих веществ.
Накопленные в XVIII столетии знания показали химикам, что судить о природе веществ, исходя только из их горючести или негорючести, нельзя. Вещества неживой природы могли выдерживать жесткую обработку, а вещества живой или некогда живой материи такой обработки не выдерживали. Вода кипела и снова конденсировалась в воду; железо или соль расплавлялись, но, остывая, возвращались в исходное состояние. В то же время оливковое масло или сахар при нагревании (даже в условиях, исключающих возможность горения) превращались в дым и гарь. То, что оставалось, не имело уже ничего общего с. оливковым маслом или сахаром, и превратить этот остаток в оливковое масло или сахар больше не удавалось. Словом, вещества этих двух групп вели себя принципиально различным образом.
В 1807 г. Берцелиус предложил вещества, подобные оливковому маслу или сахару, которые типичны для живой природы, называть органическими. Вещества, подобные воде и соли, которые характерны для неживой природы, он назвал неорганическими.
Химиков не переставало удивлять, что органические вещества при нагревании или каком-либо другом жестком воздействии легко превращаются в неорганические вещества. (Возможность обратного превращения, т. е. превращения неорганического вещества в органическое, была установлена несколько позднее.) То время было временем господства витализма — учения, рассматривающего жизнь как особое явление, подчиняющееся не законам мироздания, а влиянию особых жизненных сил (vis vitalis) [46]. Защитником витализма веком раньше был Шталь, основатель теории флогистона (см. гл. 5). Сторонники витализма утверждали, что для превращения неорганических веществ в органические требуется какое-то особое воздействие («жизненная сила»), которое проявляется только внутри живой ткани. По этой причине неорганические соединения, например воду, можно было найти повсюду — в пределах и живого, и неживого мира, тогда как органические соединения, образующиеся под воздействием жизненной силы, можно найти только в живых тканях.
Химики, имевшие дело с самыми обычными соединениями и пользовавшиеся самыми обычными методами, осуществить превращение, требовавшее участия жизненных сил, естественно, не могли.
Первые сомнения в справедливости такого утверждения возникли после опубликования в 1828 г. работы Фридриха Вёлера (1800—1882), немецкого химика, ученика Берцелиуса. Вёлера, в частности, интересовали цианиды и родственные им соединения. Нагревая цианат аммония (в то время это соединение безоговорочно причисляли к неорганическим веществам, не имеющим ничего общего с живой материей), Вёлер обнаружил, что в процессе нагревания образуются кристаллы, похожие на мочевину — продукт жизнедеятельности человека и животных, выделяющийся в значительных количествах с мочой. Тщательно изучив эти кристаллы, Вёлер установил, что он действительно получил мочевину — бесспорно органическое соединение.
Вёлер несколько раз повторил опыт и, убедившись, что он по своему желанию может превращать неорганическое соединение (цианат аммония) в органическое (мочевину), сообщил о своем открытии Берцелиусу. Берцелиус был упрямым человеком, который редко менял свое мнение под чьим-либо влиянием, однако в этом случае он вынужден был согласиться, что проведенное им, Берцелиусом, разделение на органические и неорганические соединения оказалось не таким четким, как он полагал.
Однако не надо переоценивать значения этой работы Вёлера [47]. Сама по себе она не столь уж существенна. Строго говоря, цианат аммония не является типичным неорганическим соединением, но даже если считать его таковым, то превращение цианата аммония в мочевину (как со временем и было показано) является просто результатом изменения расположения атомов внутри молекулы. И в самом деле, ведь молекула мочевины фактически является перестроенной молекулой все того же цианата аммония.
И тем не менее значение открытия Вёлера отрицать нельзя. Оно способствовало низвержению витализма [48]и вдохновило химиков на попытки синтеза органического вещества; не будь этого открытия, химики направили бы свои усилия в другом направлении.
В 1845 г. Адольф Вильгельм Герман Кольбе (1818—1884), ученик Вёлера, успешно синтезировал уксусную кислоту, считавшуюся в его время несомненно органическим веществом. Более того, он синтезировал ее таким методом, который позволил проследить всю цепь химических превращений — от исходных элементов (углерода, водорода и кислорода) до конечного продукта — уксусной кислоты. Именно такой синтез из элементов, или полный синтез, и был необходим. Если синтез мочевины Вёлера породил сомнения относительно существования «жизненной силы», то синтез уксусной кислоты Кольбе позволил решить этот вопрос.
Французский химик Пьер Эжен Марселен Бертло (1827—1907) [49] в 50-е годы XIX в. начал систематическую разработку синтеза органических соединений и достиг больших успехов. Он синтезировал, в частности, такие хорошо известные и важные соединения, как метиловый и этиловый спирты, метан, бензол, ацетилен. Бертло «нарушил границу» между неорганической и органической химией, покончив с пресловутым «запретом». В дальнейшем такое «нарушение границ» стало обычным.
Таким образом, между способностью вещества к горению и принадлежностью его к живому или неживому миру существовала определенная связь. Хотя, безусловно, были известны и исключения. Например, уголь и сера — продукты неживой материи — входили в группу горючих веществ.
Накопленные в XVIII столетии знания показали химикам, что судить о природе веществ, исходя только из их горючести или негорючести, нельзя. Вещества неживой природы могли выдерживать жесткую обработку, а вещества живой или некогда живой материи такой обработки не выдерживали. Вода кипела и снова конденсировалась в воду; железо или соль расплавлялись, но, остывая, возвращались в исходное состояние. В то же время оливковое масло или сахар при нагревании (даже в условиях, исключающих возможность горения) превращались в дым и гарь. То, что оставалось, не имело уже ничего общего с. оливковым маслом или сахаром, и превратить этот остаток в оливковое масло или сахар больше не удавалось. Словом, вещества этих двух групп вели себя принципиально различным образом.
В 1807 г. Берцелиус предложил вещества, подобные оливковому маслу или сахару, которые типичны для живой природы, называть органическими. Вещества, подобные воде и соли, которые характерны для неживой природы, он назвал неорганическими.
Химиков не переставало удивлять, что органические вещества при нагревании или каком-либо другом жестком воздействии легко превращаются в неорганические вещества. (Возможность обратного превращения, т. е. превращения неорганического вещества в органическое, была установлена несколько позднее.) То время было временем господства витализма — учения, рассматривающего жизнь как особое явление, подчиняющееся не законам мироздания, а влиянию особых жизненных сил (vis vitalis) [46]. Защитником витализма веком раньше был Шталь, основатель теории флогистона (см. гл. 5). Сторонники витализма утверждали, что для превращения неорганических веществ в органические требуется какое-то особое воздействие («жизненная сила»), которое проявляется только внутри живой ткани. По этой причине неорганические соединения, например воду, можно было найти повсюду — в пределах и живого, и неживого мира, тогда как органические соединения, образующиеся под воздействием жизненной силы, можно найти только в живых тканях.
Химики, имевшие дело с самыми обычными соединениями и пользовавшиеся самыми обычными методами, осуществить превращение, требовавшее участия жизненных сил, естественно, не могли.
Первые сомнения в справедливости такого утверждения возникли после опубликования в 1828 г. работы Фридриха Вёлера (1800—1882), немецкого химика, ученика Берцелиуса. Вёлера, в частности, интересовали цианиды и родственные им соединения. Нагревая цианат аммония (в то время это соединение безоговорочно причисляли к неорганическим веществам, не имеющим ничего общего с живой материей), Вёлер обнаружил, что в процессе нагревания образуются кристаллы, похожие на мочевину — продукт жизнедеятельности человека и животных, выделяющийся в значительных количествах с мочой. Тщательно изучив эти кристаллы, Вёлер установил, что он действительно получил мочевину — бесспорно органическое соединение.
Вёлер несколько раз повторил опыт и, убедившись, что он по своему желанию может превращать неорганическое соединение (цианат аммония) в органическое (мочевину), сообщил о своем открытии Берцелиусу. Берцелиус был упрямым человеком, который редко менял свое мнение под чьим-либо влиянием, однако в этом случае он вынужден был согласиться, что проведенное им, Берцелиусом, разделение на органические и неорганические соединения оказалось не таким четким, как он полагал.
Однако не надо переоценивать значения этой работы Вёлера [47]. Сама по себе она не столь уж существенна. Строго говоря, цианат аммония не является типичным неорганическим соединением, но даже если считать его таковым, то превращение цианата аммония в мочевину (как со временем и было показано) является просто результатом изменения расположения атомов внутри молекулы. И в самом деле, ведь молекула мочевины фактически является перестроенной молекулой все того же цианата аммония.
И тем не менее значение открытия Вёлера отрицать нельзя. Оно способствовало низвержению витализма [48]и вдохновило химиков на попытки синтеза органического вещества; не будь этого открытия, химики направили бы свои усилия в другом направлении.
В 1845 г. Адольф Вильгельм Герман Кольбе (1818—1884), ученик Вёлера, успешно синтезировал уксусную кислоту, считавшуюся в его время несомненно органическим веществом. Более того, он синтезировал ее таким методом, который позволил проследить всю цепь химических превращений — от исходных элементов (углерода, водорода и кислорода) до конечного продукта — уксусной кислоты. Именно такой синтез из элементов, или полный синтез, и был необходим. Если синтез мочевины Вёлера породил сомнения относительно существования «жизненной силы», то синтез уксусной кислоты Кольбе позволил решить этот вопрос.
Французский химик Пьер Эжен Марселен Бертло (1827—1907) [49] в 50-е годы XIX в. начал систематическую разработку синтеза органических соединений и достиг больших успехов. Он синтезировал, в частности, такие хорошо известные и важные соединения, как метиловый и этиловый спирты, метан, бензол, ацетилен. Бертло «нарушил границу» между неорганической и органической химией, покончив с пресловутым «запретом». В дальнейшем такое «нарушение границ» стало обычным.
«Кирпичики» жизни
Вёлер, Кольбе и Бертло синтезировали относительно простые органические соединения, тогда как для живой природы характерны значительно более сложные соединения типа крахмала, жиров и белков. Изучать такие соединения гораздо труднее; непросто даже установить их точный элементный состав. В целом изучение органических веществ обещало разгадку многих проблем, но подступиться к этим веществам химику прошлого века было совсем непросто.
Вначале об этих сложных соединениях было известно только то, что их можно разбить на сравнительно простые «строительные блоки» («кирпичики»), нагревая их с разбавленной кислотой или разбавленным основанием. Русский химик Константин Сигизмундович Кирхгоф (1764—1833) первым занялся детальным изучением этого вопроса. В 1812 г. ему удалось превратить крахмал, нагревая его с кислотой, в сахар, который впоследствии получил название глюкозы [50].
В 1820 г. французский химик Анри Браконно (1780—1854) таким же способом обрабатывал желатину (продукт денатурирования белка) и получил глицин— азотсодержащую органическую кислоту, относящуюся к той группе веществ, которые впоследствии были названы (Берцелиусом) аминокислотами. Глицин был первой из двадцати различных аминокислот, выделенных в следующем веке из природных белков [51].
И крахмал, и белок имеют гигантские молекулы, построенные, как выяснилось позднее, из длинных цепей, состоящих из остатков глюкозы и аминокислот соответственно. Химики XIX в. практически были лишены возможности синтезировать эти длинные цепи в лаборатории. Иначе дело обстояло с жирами.
Французский химик Мишель Эжен Шеврель (1786—1889) посвятил первую половину своей очень долгой творческой жизни изучению жиров. В 1809 г. он обработал мыло (полученное нагреванием жира со щелочью) кислотой и выделил то, что мы теперь называем жирными кислотами. Позднее он показал, что, превращаясь в мыло, жиры теряют глицерин.
Молекула глицерина сравнительно простая и построена таким образом, что к ней легко могут «прикрепиться» дополнительные группы атомов.
Следовательно, вполне логично было предположить, что, в то время как крахмал и белки, скорее всего, построены из большого числа простых остатков молекул, с жирами дело обстоит иначе. До середины XIX в. считалось, что жиры, вероятно, построены из остатков только четырех молекул: молекулы глицерина и трех молекул жирных кислот.
На этом этапе свое слово сказал Бертло. В 1854 г. он, нагревая глицерин со стеариновой кислотой (одной из самых распространенных жирных кислот, полученных из жиров), получил молекулу, состоящую из остатка молекулы глицерина и трех остатков молекул стеариновой кислоты. Этот тристеарин, который оказался идентичен тристеарину, полученному из природных жиров, был самым сложным из синтезированных к тому времени аналогов природных продуктов.
Бертло сделал еще более важный шаг. Вместо стеариновой кислоты он взял кислоты, похожие на нее, но полученные не из природных жиров, и также нагрел их с глицерином. В результате Бертло получил соединения, очень похожие на обычные жиры, но несколько отличающиеся от любого из природных жиров.
Этот синтез показал, что химик не только способен синтезировать аналоги природных продуктов, он в состоянии сделать большее. Например, он может синтезировать из продуктов неживой природы соединение, по всем своим свойствам являющееся органическим. Именно с синтезом аналогов природных продуктов связаны самые крупные достижения органической химии второй половины XIX в. (см. гл. 10).
К середине XIX в. стало уже непопулярным причислять то или иное соединение к органическим или неорганическим, исходя лишь из того, является или не является оно продуктом живой ткани. В то время уже были известны такие органические соединения, которые никак не могли быть продуктами жизнедеятельности организмов. Тем не менее деление соединений на органические и неорганические имело смысл. Свойства соединений этих классов, как выяснилось, настолько различаются, что даже приемы работы химика-органика и химика-неорганика совершенно различны.
Становилось все более очевидным, что различие между органическими и неорганическими соединениями обусловлено особенностями химического строения молекул этих соединений. Многие химики начали говорить о разных типах строения молекул органических и неорганических соединений. Молекулы большинства неорганических веществ, с которыми имели дело химики XIX в., содержат всего от двух до восьми атомов. Да и вообще в молекулах очень немногих неорганических соединений число атомов достигает десятка.
В то же время в молекулах даже простейших органических соединений содержится десять и более атомов, а нередко число атомов в молекуле органического соединения измеряется несколькими десятками. Молекулы же таких соединений, как крахмал или белок, можно без всякого преувеличения назвать гигантскими: в них насчитываются тысячи и даже сотни тысяч атомов.
Вполне понятно, что сложная органическая молекула может легко и необратимо разрушиться даже при слабом неблагоприятном воздействии, например при легком нагревании, в то время как простые неорганические молекулы не претерпевают изменений даже при жесткой обработке.
Кроме того, все без исключения органические соединения имеют в своих молекулах один или более атомов углерода. Почти все молекулы содержат также атомы водорода. Поскольку углерод и водород сами по себе горючи, то вполне можно предположить, что соединения, в которых эти элементы играют такую важную роль, также относятся к числу горючих.
Немецкий химик Фридрих Август Кекуле фон Страдонитц (1829—1886) [52]которого обычно называют Кекуле, сделал верный вывод. В учебнике, опубликованном им в 1861 г., Кекуле определил органическую химию как химию соединений углерода. Развивая эту мысль, можно определить неорганическую химию как химию соединений, не содержащих углерод. Это определение получило широкое распространение. Правда, несколько соединений углерода, в том числе диоксид углерода и карбонат кальция, скорее следует считать типичными неорганическими соединениями, чем типичными органическими. Такие соединения углерода обычно рассматриваются в трудах по неорганической химии.
Вначале об этих сложных соединениях было известно только то, что их можно разбить на сравнительно простые «строительные блоки» («кирпичики»), нагревая их с разбавленной кислотой или разбавленным основанием. Русский химик Константин Сигизмундович Кирхгоф (1764—1833) первым занялся детальным изучением этого вопроса. В 1812 г. ему удалось превратить крахмал, нагревая его с кислотой, в сахар, который впоследствии получил название глюкозы [50].
В 1820 г. французский химик Анри Браконно (1780—1854) таким же способом обрабатывал желатину (продукт денатурирования белка) и получил глицин— азотсодержащую органическую кислоту, относящуюся к той группе веществ, которые впоследствии были названы (Берцелиусом) аминокислотами. Глицин был первой из двадцати различных аминокислот, выделенных в следующем веке из природных белков [51].
И крахмал, и белок имеют гигантские молекулы, построенные, как выяснилось позднее, из длинных цепей, состоящих из остатков глюкозы и аминокислот соответственно. Химики XIX в. практически были лишены возможности синтезировать эти длинные цепи в лаборатории. Иначе дело обстояло с жирами.
Французский химик Мишель Эжен Шеврель (1786—1889) посвятил первую половину своей очень долгой творческой жизни изучению жиров. В 1809 г. он обработал мыло (полученное нагреванием жира со щелочью) кислотой и выделил то, что мы теперь называем жирными кислотами. Позднее он показал, что, превращаясь в мыло, жиры теряют глицерин.
Молекула глицерина сравнительно простая и построена таким образом, что к ней легко могут «прикрепиться» дополнительные группы атомов.
Следовательно, вполне логично было предположить, что, в то время как крахмал и белки, скорее всего, построены из большого числа простых остатков молекул, с жирами дело обстоит иначе. До середины XIX в. считалось, что жиры, вероятно, построены из остатков только четырех молекул: молекулы глицерина и трех молекул жирных кислот.
На этом этапе свое слово сказал Бертло. В 1854 г. он, нагревая глицерин со стеариновой кислотой (одной из самых распространенных жирных кислот, полученных из жиров), получил молекулу, состоящую из остатка молекулы глицерина и трех остатков молекул стеариновой кислоты. Этот тристеарин, который оказался идентичен тристеарину, полученному из природных жиров, был самым сложным из синтезированных к тому времени аналогов природных продуктов.
Бертло сделал еще более важный шаг. Вместо стеариновой кислоты он взял кислоты, похожие на нее, но полученные не из природных жиров, и также нагрел их с глицерином. В результате Бертло получил соединения, очень похожие на обычные жиры, но несколько отличающиеся от любого из природных жиров.
Этот синтез показал, что химик не только способен синтезировать аналоги природных продуктов, он в состоянии сделать большее. Например, он может синтезировать из продуктов неживой природы соединение, по всем своим свойствам являющееся органическим. Именно с синтезом аналогов природных продуктов связаны самые крупные достижения органической химии второй половины XIX в. (см. гл. 10).
К середине XIX в. стало уже непопулярным причислять то или иное соединение к органическим или неорганическим, исходя лишь из того, является или не является оно продуктом живой ткани. В то время уже были известны такие органические соединения, которые никак не могли быть продуктами жизнедеятельности организмов. Тем не менее деление соединений на органические и неорганические имело смысл. Свойства соединений этих классов, как выяснилось, настолько различаются, что даже приемы работы химика-органика и химика-неорганика совершенно различны.
Становилось все более очевидным, что различие между органическими и неорганическими соединениями обусловлено особенностями химического строения молекул этих соединений. Многие химики начали говорить о разных типах строения молекул органических и неорганических соединений. Молекулы большинства неорганических веществ, с которыми имели дело химики XIX в., содержат всего от двух до восьми атомов. Да и вообще в молекулах очень немногих неорганических соединений число атомов достигает десятка.
В то же время в молекулах даже простейших органических соединений содержится десять и более атомов, а нередко число атомов в молекуле органического соединения измеряется несколькими десятками. Молекулы же таких соединений, как крахмал или белок, можно без всякого преувеличения назвать гигантскими: в них насчитываются тысячи и даже сотни тысяч атомов.
Вполне понятно, что сложная органическая молекула может легко и необратимо разрушиться даже при слабом неблагоприятном воздействии, например при легком нагревании, в то время как простые неорганические молекулы не претерпевают изменений даже при жесткой обработке.
Кроме того, все без исключения органические соединения имеют в своих молекулах один или более атомов углерода. Почти все молекулы содержат также атомы водорода. Поскольку углерод и водород сами по себе горючи, то вполне можно предположить, что соединения, в которых эти элементы играют такую важную роль, также относятся к числу горючих.
Немецкий химик Фридрих Август Кекуле фон Страдонитц (1829—1886) [52]которого обычно называют Кекуле, сделал верный вывод. В учебнике, опубликованном им в 1861 г., Кекуле определил органическую химию как химию соединений углерода. Развивая эту мысль, можно определить неорганическую химию как химию соединений, не содержащих углерод. Это определение получило широкое распространение. Правда, несколько соединений углерода, в том числе диоксид углерода и карбонат кальция, скорее следует считать типичными неорганическими соединениями, чем типичными органическими. Такие соединения углерода обычно рассматриваются в трудах по неорганической химии.