Фрактальное пространство-время генома, пангеномы и кластеризация прокариот
В главе 3 мы сосредоточились на трехкомпонентной структуре прокариотического геномного пространства, состоящего из ядра, оболочки и облака, и показали, что эта структура фрактальна. Одни и те же три компонента, а именно небольшое ядро, оболочка большего размера и огромное по сравнению с ними «облако», проявляются на любом уровне разбиения генного пространства, от мира прокариот в целом до совсем небольших групп бактерий (см. рис. 3-14). Непосредственным следствием этой фрактальности является важность «пангеномов» – всей общности генов, представляющих геномы, принадлежащие к кластеру архей или бактерий на данном уровне. Читатель может (и должен) немедленно спросить, что определяет кластеры и откуда берутся уровни. Пока предположим, что дерево рРНК Карла Вёзе (см. рис. 2–3) разумно описывает организацию пространства-времени мира прокариот и является по крайней мере одним из источников для кластеризации. В главе 6 мы обсудим применимость и смысл концепции древа жизни более глубоко и покажем, что дерево рРНК, хотя ни в коем случае и не является полным представлением истории эволюции прокариот, тем не менее вполне осмысленно.
Огромное множество архейных и бактериальных генов кодируют белки, которые не имеют никакого измеримого сходства с какими-либо другими доступными последовательностями белков. Эти гены часто обозначают как одинокие рамки считывания (ОРС)[50] (Daubin and Ochman, 2004). Обычно в архейных и бактериальных геномах ОРС составляют 10–15 процентов от всех предсказанных генов. Многие ОРС – очень короткие, и некоторые из них могут быть не реальными генами, а результатом ошибочного предсказания при анализе генома (Ochman, 2002). Кроме того, высказывается предположение, что большинство ОРС, являющихся полноценными генами, произошли от генов бактериофагов и, соответственно, характеризуются высокой горизонтальной мобильностью, хотя в некоторых случаях они могут быть задействованы для клеточных функций и, соответственно, фиксируются в бактериальных и архейных геномах. Последние оценки, следующие из метагеномных исследований бактериофагов, предполагают, что разнообразие фаговых последовательностей очень велико и остается по большей части неизученным (Edwards and Rohwer, 2005). Таким образом, кажется привлекательной идея, что бо́льшая часть бактериальных и архейных ОРС произошла из этого огромного резервуара генов. В трехкомпонентной структуре вселенной прокариотических генов, с которой мы теперь знакомы, ОРС естественным образом объединяются с «облаком» редких генов, которые количественно доминируют в генном пространстве, но не в индивидуальных геномах, как обсуждалось в главе 3.
Насколько велико все геномное пространство прокариот? Сколько генов в общей сложности оно содержит? Надежная экстраполяция расширения геномного пространства в результате продолжающегося секвенирования бактериальных и архейных геномов и достоверная оценка реального размера этого пространства трудноосуществимы. Однако с учетом большого разнообразия микробных виромов, которые являются основным резервуаром генов и их переносчиком (см. также гл. 10), наиболее вероятно, что число элементов прокариотического геномного пространства увеличится на порядки величины, в основном, если не исключительно, за счет расширения «облака» (Koonin and Wolf, 2008b; Lapierre and Gogarten, 2009).
Огромное множество архейных и бактериальных генов кодируют белки, которые не имеют никакого измеримого сходства с какими-либо другими доступными последовательностями белков. Эти гены часто обозначают как одинокие рамки считывания (ОРС)[50] (Daubin and Ochman, 2004). Обычно в архейных и бактериальных геномах ОРС составляют 10–15 процентов от всех предсказанных генов. Многие ОРС – очень короткие, и некоторые из них могут быть не реальными генами, а результатом ошибочного предсказания при анализе генома (Ochman, 2002). Кроме того, высказывается предположение, что большинство ОРС, являющихся полноценными генами, произошли от генов бактериофагов и, соответственно, характеризуются высокой горизонтальной мобильностью, хотя в некоторых случаях они могут быть задействованы для клеточных функций и, соответственно, фиксируются в бактериальных и архейных геномах. Последние оценки, следующие из метагеномных исследований бактериофагов, предполагают, что разнообразие фаговых последовательностей очень велико и остается по большей части неизученным (Edwards and Rohwer, 2005). Таким образом, кажется привлекательной идея, что бо́льшая часть бактериальных и архейных ОРС произошла из этого огромного резервуара генов. В трехкомпонентной структуре вселенной прокариотических генов, с которой мы теперь знакомы, ОРС естественным образом объединяются с «облаком» редких генов, которые количественно доминируют в генном пространстве, но не в индивидуальных геномах, как обсуждалось в главе 3.
Насколько велико все геномное пространство прокариот? Сколько генов в общей сложности оно содержит? Надежная экстраполяция расширения геномного пространства в результате продолжающегося секвенирования бактериальных и архейных геномов и достоверная оценка реального размера этого пространства трудноосуществимы. Однако с учетом большого разнообразия микробных виромов, которые являются основным резервуаром генов и их переносчиком (см. также гл. 10), наиболее вероятно, что число элементов прокариотического геномного пространства увеличится на порядки величины, в основном, если не исключительно, за счет расширения «облака» (Koonin and Wolf, 2008b; Lapierre and Gogarten, 2009).
Эволюционная динамика архитектуры генома прокариот: опероны, суперопероны и сети соседствующих генов
Как уже отмечалось в главе 3, практически сразу же, как только были опубликованы первые полные геномные последовательности, стало очевидным, что последовательность генов в бактериальных и архейных геномах относительно мало консервативна, она сохраняется существенно хуже, чем последовательность нуклеотидов в самих генах (см. рис. 3–6). Для того чтобы анализировать эволюцию последовательности генов, необходимо иметь надежный набор ортологичных генов в сравниваемых геномах (см. табл. 3–1). Как только такое множество ортологичных генов задано, становится достаточно просто оценить степень сохранения последовательности генов, например с помощью точечного графика (одно из самых ранних представлений степени сходства нуклеотидных и белковых последовательностей), в котором каждая точка представляет собой пару ортологов. Исследование этих графиков показывает быстрое расхождение порядка генов у прокариот таким образом, что даже между близкородственными организмами коллинеарность хромосом разрушена в нескольких точках (см. рис. 5–2а), a умеренно разошедшиеся организмы показывают лишь несколько протяженных коллинеарных районов (см. рис. 5–2б и 5–2в). Для любой пары более отдаленных друг от друга организмов график выглядит как карта звездного неба (см. рис. 5–2 г). Разрушение синтении в процессе эволюции бактериальных и архейных геномов обычно явно бросается в глаза на графике, образуя картину в форме буквы X (см. рис. 5–2б и 5–2в). В свое время было сделано предположение, что такая картина возникает в результате симметричных хромосомных инверсий с центром в точке начала репликации (Eisen et al., 2000). Исходной причиной таких инверсий может быть высокая частота рекомбинаций в репликационных вилках, которые в кольцевых хромосомах бактерий и архей обычно располагаются с обеих сторон и на одинаковом расстоянии от точки начала репликации.
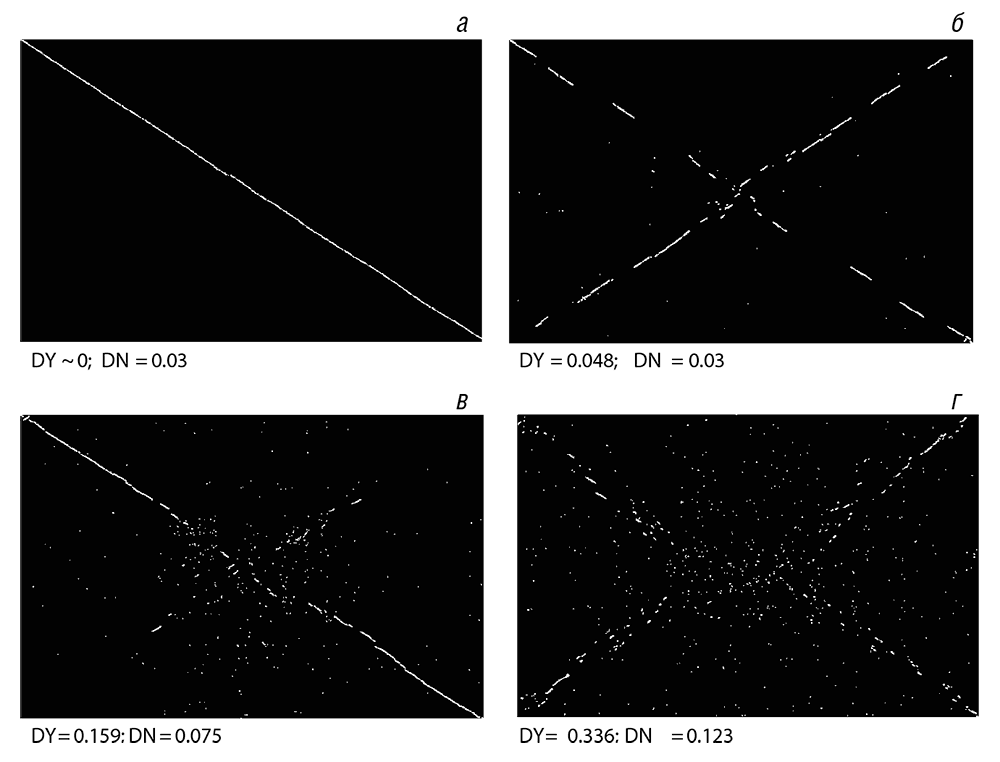 Рис. 5–2. Расхождение порядка следования генов между геномами бактерий: а – Borrelia afzelii PKo по сравнению с Borrelia burgdorferi B31; б – Shewanella oneidensis MR-1 по сравнению с Shewanella sp. ANA-3; в – Pseudomonas fluorescens PfO-1 по сравнению с Pseudomonas fluorescens Pf-5; г – Pseudomonas fluorescens Pf-5 по сравнению с Pseudomonas syringae pv. tomato str. DC3000. Каждая точка представляет пару ортологичных генов, идентифицированных с использованием метода наилучшего сходства при двунаправленном сравнении (см. табл. 3–1). Яркие точки показывают пары ортологичных генов, принадлежащих консервативным массивам генов; бледные точки показывают изолированные ортологи. DY – расстояние между сравниваемыми геномами в терминах порядка следования генов, как описано в Novichkov et al., 2009. DN – медианное расстояние между последовательностями несинонимических сайтов в белок-кодирующих генах.
Рис. 5–2. Расхождение порядка следования генов между геномами бактерий: а – Borrelia afzelii PKo по сравнению с Borrelia burgdorferi B31; б – Shewanella oneidensis MR-1 по сравнению с Shewanella sp. ANA-3; в – Pseudomonas fluorescens PfO-1 по сравнению с Pseudomonas fluorescens Pf-5; г – Pseudomonas fluorescens Pf-5 по сравнению с Pseudomonas syringae pv. tomato str. DC3000. Каждая точка представляет пару ортологичных генов, идентифицированных с использованием метода наилучшего сходства при двунаправленном сравнении (см. табл. 3–1). Яркие точки показывают пары ортологичных генов, принадлежащих консервативным массивам генов; бледные точки показывают изолированные ортологи. DY – расстояние между сравниваемыми геномами в терминах порядка следования генов, как описано в Novichkov et al., 2009. DN – медианное расстояние между последовательностями несинонимических сайтов в белок-кодирующих генах.
Одной из наиболее ранних фундаментальных концепций бактериальной генетики является оперон, то есть группа совместно транскрибируемых и регулируемых генов (Jacob and Monod, 1961). Гипотеза оперона – выдающийся концептуальный прорыв Франсуа Жакоба и Жака Моно. Хотя за 50 лет, прошедших с момента ее первой публикации, было открыто огромное количество вариаций простой схемы регуляции лактозного оперона Lac репрессором, оперон выдержал проверку сравнительной геномикой как главный организационный принцип бактериальных и архейных геномов. В процессе эволюции опероны сохраняются гораздо лучше, чем протяженные синтении. Тем не менее сравнительный анализ порядка следования генов в бактериях и археях выявил небольшое количество оперонов, общих для широкого многообразия организмов. Как уже было отмечено ранее, высококонсервативные опероны, как правило, кодируют физически взаимодействующие белки, тенденция, легко объяснимая отбором, направленным против вредных эффектов дисбаланса между субъединицами сложных белковых комплексов. Наиболее эффектной иллюстрацией этой тенденции является рибосомный супероперон, включающий более 50 генов рибосомных белков, который встречается в различных комбинациях и локализациях во всех секвенированных архейных и бактериальных геномах. Анализ рибосомного супероперона и других частично сохраняющихся групп оперонов меньшего размера привел к идее сверхоперона (Lathe et al., 2000), или консервативного окружения гена (Rogozin et al., 2002), как некоего множества перекрывающихся, частично консервативных цепочек генов (известных или предсказанных оперонов; см. рис. 5–3). В дополнение к рибосомному супероперону, яркими примерами консервативного окружения являются предсказанная группа перекрывающихся оперонов, которая кодирует субъединицы экзосомного комплекса архей, и cas-гены, из которых состоит антивирусная система защиты (см. также гл. 9 и 10).
Большинство генов в каждом консервативном окружении кодируют белки, вовлеченные в один и тот же процесс или комплекс, но существуют и высококонсервативные участки, которые включают гены с функциями, как кажется, несвязанными. Яркий пример – частое присутствие гена енолазы в рибосомном окружении или генов субъединиц протеасомы в экзосомном окружении архей. Присутствие этих генов, на первый взгляд кажущихся неуместными в консервативном генном окружении, может объясняться скрытой функциональной связью, плейотропией (множественностью функций соответствующих белков), или «геномным автостопом», когда оперон объединяет гены функционально не связанные, но экспрессируемые в одинаковых условиях (Rogozin et al., 2002).
Концепция геномного окружения воплощает в себе парадигму эволюции генома прокариот, если не эволюции геномов вообще, так как она ярко демонстрирует баланс между частичным сохранением элементов ядра и огромной диверсификацией периферии (см. рис. 5–3а). Так же как для многих других объектов и их взаимоотношений в биологии, эти частично консервативные окружения могут быть естественным образом представлены в виде сети, в которой гены являются узлами, соседи соединены ребрами, а вес ребер пропорционален частоте встречаемости данной связи в геномах (см. рис. 5–3).
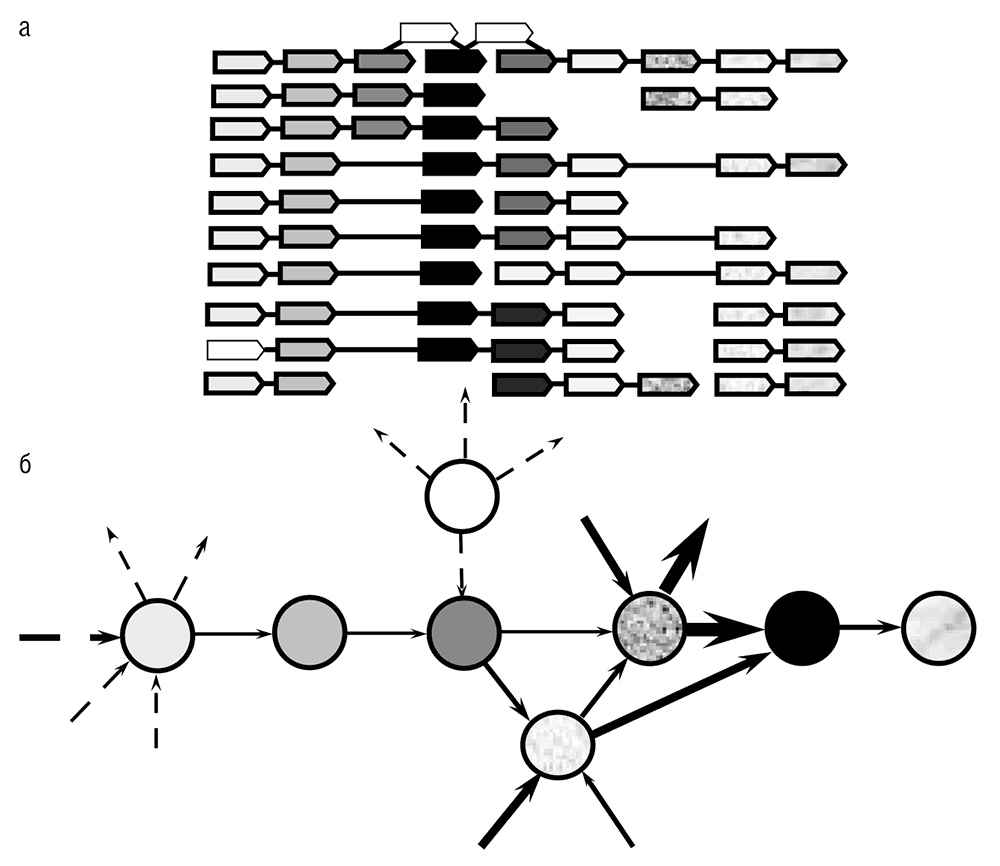 Рис. 5–3. Частично сохраняющееся окружение гена в геноме прокариот: а – перекрывающиеся, частично сохраняющиеся массивы генов. Гены показаны в форме стрелок с уникальной штриховкой или текстурой. Связывающие их жирные линии обозначают короткие межгенные промежутки, а тонкие линии показывают протяженные районы, разделяющие соответствующие гены. (Они содержат дополнительные гены и изображены без учета масштаба.) В случаях, в которых опероны не связаны, они могут располагаться в различных частях генома. На рисунке показаны реальные массивы генов, но названия конкретных геномов и генов не указаны, чтобы подчеркнуть общий характер геномной организации подобного типа. Данные по Rogozin et al., 2002; б – представление окружения гена в виде сети. Закрашенные окружности показывают гены, которые принадлежат к окружению, автоматически вычлененному с использованием алгоритма, описанного в Rogozin et al., 2002; показана только часть окружения. Незакрашенная окружность соответствует гену, который принадлежит окружению, но не был включен в него автоматической процедурой. Стрелки показывают связь между генами в оперонах (жирные стрелки соответствуют связям внутри окружения, а пунктирные стрелки – внешним связям). Толщина стрелок примерно пропорциональна числу геномов, в которых представлена данная пара генов.
Рис. 5–3. Частично сохраняющееся окружение гена в геноме прокариот: а – перекрывающиеся, частично сохраняющиеся массивы генов. Гены показаны в форме стрелок с уникальной штриховкой или текстурой. Связывающие их жирные линии обозначают короткие межгенные промежутки, а тонкие линии показывают протяженные районы, разделяющие соответствующие гены. (Они содержат дополнительные гены и изображены без учета масштаба.) В случаях, в которых опероны не связаны, они могут располагаться в различных частях генома. На рисунке показаны реальные массивы генов, но названия конкретных геномов и генов не указаны, чтобы подчеркнуть общий характер геномной организации подобного типа. Данные по Rogozin et al., 2002; б – представление окружения гена в виде сети. Закрашенные окружности показывают гены, которые принадлежат к окружению, автоматически вычлененному с использованием алгоритма, описанного в Rogozin et al., 2002; показана только часть окружения. Незакрашенная окружность соответствует гену, который принадлежит окружению, но не был включен в него автоматической процедурой. Стрелки показывают связь между генами в оперонах (жирные стрелки соответствуют связям внутри окружения, а пунктирные стрелки – внешним связям). Толщина стрелок примерно пропорциональна числу геномов, в которых представлена данная пара генов.
Большинство оперонов находится не в сложном окружении, включающем разнообразные связи, а представляет собой простую последовательность от двух до четырех генов, порядок которых может различаться. Идентичные или похожие в смысле организации генов опероны часто обнаруживаются в сильно различающихся организмах и в различных функциональных системах. Примечательны в данном случае многочисленные опероны транспорта метаболитов, которые состоят из расположенных в одинаковом порядке генов, кодирующих трансмембранные пермеазы, АТФазы и периплазматические субъединицы так называемых ABC-транспортеров (три субъединицы обозначаются соответственно A, B и C). Присутствие таких общих оперонов в разнообразных бактериях и археях было интерпретировано в рамках гипотезы эгоистичного оперона (Lawrence, 1999), которая постулирует, что оперон так хорошо сохраняется не из-за функциональной важности совместной регуляции входящих в него генов, а из-за «эгоистичности» этой компактной генетической единицы, которая склонна к горизонтальному распространению среди прокариот (ниже в этой главе мы еще вернемся к данной концепции при обсуждении горизонтального переноса генов).
Систематическое сравнение расположения ортологичных генов в архейных и бактериальных геномах выявило относительно небольшую долю сохраняющихся (предсказанных) оперонов и гораздо большую распространенность уникальных директонов (последовательностей генов, считываемых в одинаковом направлении и разделенных короткими межгенными участками; Wolf et al., 2001). Как было показано, возможно несколько неожиданно, директоны довольно точно предсказывают опероны: большинство директонов в действительности, по-видимому, являются оперонами (Salgado et al., 2000). Таким образом, архейные и бактериальные геномы сформированы на оперонных принципах с небольшим числом высококонсервативных оперонов и намного более многочисленными редкими и уникальными оперонами. С учетом этого обстоятельства модель консервации оперонов, по крайней мере качественно, напоминает распределение кластеров ортологичных генов, с его трехкомпонентной структурой (см. выше): редкие гены и редкие опероны гораздо более многочисленны, чем повсеместно распространенные гены и опероны.
Степень «оперонизации» генома у бактерий и архей широко варьирует: некоторые геномы, например как у гипертермофильной бактерии Thermotoga maritima, почти полностью состоят из (предсказанных) оперонов, в то время как другие, как у большинства цианобактерий, по-видимому, содержат очень немного оперонов. Остается неясным, что определяет распространенность оперонов в организме, хотя высказывались предположения, что степень «оперонизации» зависит от баланса между интенсивностью рекомбинации и горизонтального потока генов, а также факторов отбора, препятствующих разрушению оперонов.
Одной из наиболее ранних фундаментальных концепций бактериальной генетики является оперон, то есть группа совместно транскрибируемых и регулируемых генов (Jacob and Monod, 1961). Гипотеза оперона – выдающийся концептуальный прорыв Франсуа Жакоба и Жака Моно. Хотя за 50 лет, прошедших с момента ее первой публикации, было открыто огромное количество вариаций простой схемы регуляции лактозного оперона Lac репрессором, оперон выдержал проверку сравнительной геномикой как главный организационный принцип бактериальных и архейных геномов. В процессе эволюции опероны сохраняются гораздо лучше, чем протяженные синтении. Тем не менее сравнительный анализ порядка следования генов в бактериях и археях выявил небольшое количество оперонов, общих для широкого многообразия организмов. Как уже было отмечено ранее, высококонсервативные опероны, как правило, кодируют физически взаимодействующие белки, тенденция, легко объяснимая отбором, направленным против вредных эффектов дисбаланса между субъединицами сложных белковых комплексов. Наиболее эффектной иллюстрацией этой тенденции является рибосомный супероперон, включающий более 50 генов рибосомных белков, который встречается в различных комбинациях и локализациях во всех секвенированных архейных и бактериальных геномах. Анализ рибосомного супероперона и других частично сохраняющихся групп оперонов меньшего размера привел к идее сверхоперона (Lathe et al., 2000), или консервативного окружения гена (Rogozin et al., 2002), как некоего множества перекрывающихся, частично консервативных цепочек генов (известных или предсказанных оперонов; см. рис. 5–3). В дополнение к рибосомному супероперону, яркими примерами консервативного окружения являются предсказанная группа перекрывающихся оперонов, которая кодирует субъединицы экзосомного комплекса архей, и cas-гены, из которых состоит антивирусная система защиты (см. также гл. 9 и 10).
Большинство генов в каждом консервативном окружении кодируют белки, вовлеченные в один и тот же процесс или комплекс, но существуют и высококонсервативные участки, которые включают гены с функциями, как кажется, несвязанными. Яркий пример – частое присутствие гена енолазы в рибосомном окружении или генов субъединиц протеасомы в экзосомном окружении архей. Присутствие этих генов, на первый взгляд кажущихся неуместными в консервативном генном окружении, может объясняться скрытой функциональной связью, плейотропией (множественностью функций соответствующих белков), или «геномным автостопом», когда оперон объединяет гены функционально не связанные, но экспрессируемые в одинаковых условиях (Rogozin et al., 2002).
Концепция геномного окружения воплощает в себе парадигму эволюции генома прокариот, если не эволюции геномов вообще, так как она ярко демонстрирует баланс между частичным сохранением элементов ядра и огромной диверсификацией периферии (см. рис. 5–3а). Так же как для многих других объектов и их взаимоотношений в биологии, эти частично консервативные окружения могут быть естественным образом представлены в виде сети, в которой гены являются узлами, соседи соединены ребрами, а вес ребер пропорционален частоте встречаемости данной связи в геномах (см. рис. 5–3).
Большинство оперонов находится не в сложном окружении, включающем разнообразные связи, а представляет собой простую последовательность от двух до четырех генов, порядок которых может различаться. Идентичные или похожие в смысле организации генов опероны часто обнаруживаются в сильно различающихся организмах и в различных функциональных системах. Примечательны в данном случае многочисленные опероны транспорта метаболитов, которые состоят из расположенных в одинаковом порядке генов, кодирующих трансмембранные пермеазы, АТФазы и периплазматические субъединицы так называемых ABC-транспортеров (три субъединицы обозначаются соответственно A, B и C). Присутствие таких общих оперонов в разнообразных бактериях и археях было интерпретировано в рамках гипотезы эгоистичного оперона (Lawrence, 1999), которая постулирует, что оперон так хорошо сохраняется не из-за функциональной важности совместной регуляции входящих в него генов, а из-за «эгоистичности» этой компактной генетической единицы, которая склонна к горизонтальному распространению среди прокариот (ниже в этой главе мы еще вернемся к данной концепции при обсуждении горизонтального переноса генов).
Систематическое сравнение расположения ортологичных генов в архейных и бактериальных геномах выявило относительно небольшую долю сохраняющихся (предсказанных) оперонов и гораздо большую распространенность уникальных директонов (последовательностей генов, считываемых в одинаковом направлении и разделенных короткими межгенными участками; Wolf et al., 2001). Как было показано, возможно несколько неожиданно, директоны довольно точно предсказывают опероны: большинство директонов в действительности, по-видимому, являются оперонами (Salgado et al., 2000). Таким образом, архейные и бактериальные геномы сформированы на оперонных принципах с небольшим числом высококонсервативных оперонов и намного более многочисленными редкими и уникальными оперонами. С учетом этого обстоятельства модель консервации оперонов, по крайней мере качественно, напоминает распределение кластеров ортологичных генов, с его трехкомпонентной структурой (см. выше): редкие гены и редкие опероны гораздо более многочисленны, чем повсеместно распространенные гены и опероны.
Степень «оперонизации» генома у бактерий и архей широко варьирует: некоторые геномы, например как у гипертермофильной бактерии Thermotoga maritima, почти полностью состоят из (предсказанных) оперонов, в то время как другие, как у большинства цианобактерий, по-видимому, содержат очень немного оперонов. Остается неясным, что определяет распространенность оперонов в организме, хотя высказывались предположения, что степень «оперонизации» зависит от баланса между интенсивностью рекомбинации и горизонтального потока генов, а также факторов отбора, препятствующих разрушению оперонов.
Регуляция экспрессии генов и передачи сигналов у бактерий и архей: от базовой схемы оперона к сверхоперонам, регулонам и сложным сетям
Бактерии и археи обладают сложной и элегантной системой регуляции экспрессии генов. Сравнительная геномика драматически изменила существующие взгляды на принципы организации, распределение в природе и эволюцию этих регуляторных механизмов. Концепция оперона Жакоба и Моно, представленная в предыдущем разделе как основной принцип локальной архитектуры бактериальных и архейных геномов, также является концепцией регуляции экспрессии генов и передачи сигналов у прокариот. В модели Жакоба – Моно регулятор (репрессор лактозы в их оригинальной работе) является сенсором внеклеточных и внутриклеточных сигналов (в данном случае концентрации лактозы), что влияет на структуру белка-регулятора и, опосредованно, на экспрессию оперона (в случае лактозного оперона репрессор, связывая лактозу, отсоединяется от регуляторной части оперона, делая тем самым возможной транскрипцию). В течение полувека, прошедших с момента фундаментального открытия Жакоба – Моно, было обнаружено множество вариаций этой темы, включая регуляторы, которые симметрично влияют на транскрипцию разных расположенных по соседству генов, и глобальные регуляторы, которые контролируют экспрессию многочисленных разрозненных генов и оперонов, в противоположность репрессору простого оперона в модели Жакоба – Моно. Наиболее заметными глобальными регуляторами являются белки – подавители катаболизма (CRP) и регулятор ответа на стресс (SOS) LexA. С учетом открытия этих и других глобальных регуляторов концепция оперона была усовершенствована понятием регулона – набора генов, экспрессия которых регулируется одним и тем же белком-регулятором. Сравнительный геномный анализ регулонов выявил их чрезвычайную эволюционную пластичность с существенными различиями между регулонами даже у близкородственных организмов (Lozada-Chavez et al., 2006). Глобальные регуляторы транскрипции, такие как LexA, широко распространены и высококонсервативны в различных бактериях, но состав генов в регулоне LexA является очень вариативным. Пластичность регулонов, наряду с изменчивостью архитектуры генома (см. выше), хорошо согласуется с идеей, что регуляция экспрессии генов и архитектура генома в эволюции архей и бактерий тесно взаимосвязаны. В резком контрасте с изменчивостью и пластичностью регулонов, регуляторы транскрипции у бактерий и архей демонстрируют примечательное единство архитектуры и структуры. Как правило, эти регуляторы содержат домен, связывающий небольшие молекулы-сенсоры и ДНК-связывающий домен. Подавляющее большинство ДНК-связывающих доменов являются вариациями одной и той же структурной темы, спираль – поворот – спираль. Более специфические, но тоже распространенные домены связывания с ДНК включают мотивы лента – спираль – спираль и цинковая лента (Aravind et al., 2005; Aravind and Koonin, 1999).
Более сложная схема передачи сигналов и регуляции экспрессии генов, которая процессирует сигналы, приходящие из окружающей среды, основана на так называемых двухкомпонентных системах (Casino et al., 2010). Двухкомпонентные системы состоят из мембранных гистидин-киназ и растворимых регуляторов ответа, между которыми сигнал передается путем переноса фосфата. Примечательно, что классические регуляторы транскрипции и гистидин-киназы содержат много общих сенсорных доменов. Это родство указывает на то, что регуляторы транскрипции (однокомпонентные системы) и двухкомпонентные системы образуют единую, интегрированную структуру передачи сигналов и регуляции экспрессии. Однокомпонентные системы, которые распространены практически повсеместно и, как правило, численно доминируют у бактерий и архей, предположительно являются наиболее древними устройствами передачи сигналов, в то время как двухкомпонентные системы, вероятно, являются произошедшей от них более сложной формой передачи сигнала, которая эволюционировала как механизм реагирования на стимулы, приходящие из окружающей среды (Ulrich et al., 2005).
Сравнительная геномика бактерий и архей внесла решающий вклад в открытие новых, до того неизвестных, но в действительности весьма распространенных систем передачи сигналов. В течение многих лет было известно, что широко распространенная форма глобальной регуляции у бактерий использует в качестве посредника цАМФ (циклический АМФ), при участии различных аденилатциклаз (яркий пример неортологичной замены генов), многочисленных белков, содержащих сенсоры цАМФ, такие как GAF-домен, a также белки катаболитной репрессии (CRP и FNR) и другие регуляторы транскрипции, которые тоже содержат цАМФ-связывающие домены. Сравнительный анализ выявил многочисленные неклассифицированные белки, содержащие гомологичные сенсорные домены, которые типичны для цАМФ-зависимых регуляторов и двухкомпонентных систем, объединенные с одним или двумя новыми доменами, GGDEF и EAL (обозначенными так по соответствующим мотивам консервативных последовательностей аминокислот). Геномный контекст этих доменов и наблюдение, что домен GGDEF является отдаленным гомологом одного из семейств аденилатциклаз, привели к гипотезе, что эти белки являются компонентами новой системы (или систем) передачи сигналов. Впоследствии эти предсказанные системы были открыты после того, как было показано, что домен GGDEF обладает активностью ди-ГМФ-циклазы, в то время как EAL является ди-ГМФ-фосфодиэстеразой. Зависимая от ц-ди-ГМФ передача сигнала, существование которой даже не предполагалось в догеномную эру, начинает рассматриваться как главная регуляторная система бактерий и архей (Seshasayee et al., 2010).
Другая интересная тема дискуссий – широкое представительство у прокариот различных модулей сложных систем передачи сигналов, которые, как считалось ранее, характерны только для эукариот. В частности, сравнительный геномный анализ убедительно показал, что белковые серин-треонин-киназы и соответствующие фосфатазы широко распространены и диверсифицированы среди архей и бактерий и являются важным компонентом многогранной системы передачи сигналов у прокариот. Анализ большего количества бактериальных геномов неожиданно выявил гомологи белков, которые, как считалось ранее, имеются только у эукариот, где они вовлечены в известные пути передачи сигналов, такие как программируемая клеточная смерть (ПКС), или апоптоз. Эти белки включают протеазы из суперсемейства каспаз, семейство апоптозных АТФаз и семейство ГТФаз NACHT; все они вовлечены в различные формы ПКС растений и животных (Koonin and Aravind, 2002; Leipe et al., 2004). Как правило, эти белки обладают сложной мультидоменной модульной архитектурой, для которой характерно соединение каталитических доменов с разнообразными доменами, обеспечивающими специфичность белок-белковых взаимодействий. Эти предполагаемые сигнальные молекулы наиболее распространены в бактериях со сложными фазами развития, таких как цианобактерии, актинобактерии и миксобактерии, а также присутствуют у метаносарцин, единственной известной группы архей с относительно большими геномами и сложной морфологией. Детальное исследование функций этих белков еще предстоит, но есть предварительные признаки того, что у некоторых бактерий они могут быть вовлечены в ПКС (Bidle and Falkowski, 2004). Эти наблюдения показывают, что по крайней мере для некоторых из сложных сигнальных систем эукариот существуют аналоги и вероятные эволюционные предшественники среди бактерий. Мы еще вернемся к этим связям, когда будем обсуждать в главе 7 происхождение эукариот.
Наряду с вышеупомянутой приблизительно квадратичной зависимостью от размера генома сравнительный геномный анализ выявил огромную вариацию в сложности систем передачи сигналов среди бактерий и архей. Эта изменчивость, по-видимому, отражает разнообразие стилей жизни среди соответствующих организмов. Вариации в доле генов, ответственных за передачу сигналов, были количественно отражены в «бактериальном IQ», показателе, который пропорционален квадратному корню от числа белков передачи сигналов (учитывая квадратичное масштабирование) и обратно пропорционален общему количеству генов (Galperin, 2005). IQ отражает способность бактерий и архей отвечать на различные стимулы, приходящие из внешней среды. Соответственно, внутриклеточные симбионты (паразиты) имеют наименьшие значения IQ. Он лишь ненамного выше у организмов с компактными геномами, живущих в стабильной внешней середе, таких как морские цианобактерии, и существенно больше у организмов, живущих в сложной и переменчивой среде, даже у тех, которые обладают сравнительно небольшими геномами.
Более сложная схема передачи сигналов и регуляции экспрессии генов, которая процессирует сигналы, приходящие из окружающей среды, основана на так называемых двухкомпонентных системах (Casino et al., 2010). Двухкомпонентные системы состоят из мембранных гистидин-киназ и растворимых регуляторов ответа, между которыми сигнал передается путем переноса фосфата. Примечательно, что классические регуляторы транскрипции и гистидин-киназы содержат много общих сенсорных доменов. Это родство указывает на то, что регуляторы транскрипции (однокомпонентные системы) и двухкомпонентные системы образуют единую, интегрированную структуру передачи сигналов и регуляции экспрессии. Однокомпонентные системы, которые распространены практически повсеместно и, как правило, численно доминируют у бактерий и архей, предположительно являются наиболее древними устройствами передачи сигналов, в то время как двухкомпонентные системы, вероятно, являются произошедшей от них более сложной формой передачи сигнала, которая эволюционировала как механизм реагирования на стимулы, приходящие из окружающей среды (Ulrich et al., 2005).
Сравнительная геномика бактерий и архей внесла решающий вклад в открытие новых, до того неизвестных, но в действительности весьма распространенных систем передачи сигналов. В течение многих лет было известно, что широко распространенная форма глобальной регуляции у бактерий использует в качестве посредника цАМФ (циклический АМФ), при участии различных аденилатциклаз (яркий пример неортологичной замены генов), многочисленных белков, содержащих сенсоры цАМФ, такие как GAF-домен, a также белки катаболитной репрессии (CRP и FNR) и другие регуляторы транскрипции, которые тоже содержат цАМФ-связывающие домены. Сравнительный анализ выявил многочисленные неклассифицированные белки, содержащие гомологичные сенсорные домены, которые типичны для цАМФ-зависимых регуляторов и двухкомпонентных систем, объединенные с одним или двумя новыми доменами, GGDEF и EAL (обозначенными так по соответствующим мотивам консервативных последовательностей аминокислот). Геномный контекст этих доменов и наблюдение, что домен GGDEF является отдаленным гомологом одного из семейств аденилатциклаз, привели к гипотезе, что эти белки являются компонентами новой системы (или систем) передачи сигналов. Впоследствии эти предсказанные системы были открыты после того, как было показано, что домен GGDEF обладает активностью ди-ГМФ-циклазы, в то время как EAL является ди-ГМФ-фосфодиэстеразой. Зависимая от ц-ди-ГМФ передача сигнала, существование которой даже не предполагалось в догеномную эру, начинает рассматриваться как главная регуляторная система бактерий и архей (Seshasayee et al., 2010).
Другая интересная тема дискуссий – широкое представительство у прокариот различных модулей сложных систем передачи сигналов, которые, как считалось ранее, характерны только для эукариот. В частности, сравнительный геномный анализ убедительно показал, что белковые серин-треонин-киназы и соответствующие фосфатазы широко распространены и диверсифицированы среди архей и бактерий и являются важным компонентом многогранной системы передачи сигналов у прокариот. Анализ большего количества бактериальных геномов неожиданно выявил гомологи белков, которые, как считалось ранее, имеются только у эукариот, где они вовлечены в известные пути передачи сигналов, такие как программируемая клеточная смерть (ПКС), или апоптоз. Эти белки включают протеазы из суперсемейства каспаз, семейство апоптозных АТФаз и семейство ГТФаз NACHT; все они вовлечены в различные формы ПКС растений и животных (Koonin and Aravind, 2002; Leipe et al., 2004). Как правило, эти белки обладают сложной мультидоменной модульной архитектурой, для которой характерно соединение каталитических доменов с разнообразными доменами, обеспечивающими специфичность белок-белковых взаимодействий. Эти предполагаемые сигнальные молекулы наиболее распространены в бактериях со сложными фазами развития, таких как цианобактерии, актинобактерии и миксобактерии, а также присутствуют у метаносарцин, единственной известной группы архей с относительно большими геномами и сложной морфологией. Детальное исследование функций этих белков еще предстоит, но есть предварительные признаки того, что у некоторых бактерий они могут быть вовлечены в ПКС (Bidle and Falkowski, 2004). Эти наблюдения показывают, что по крайней мере для некоторых из сложных сигнальных систем эукариот существуют аналоги и вероятные эволюционные предшественники среди бактерий. Мы еще вернемся к этим связям, когда будем обсуждать в главе 7 происхождение эукариот.
Наряду с вышеупомянутой приблизительно квадратичной зависимостью от размера генома сравнительный геномный анализ выявил огромную вариацию в сложности систем передачи сигналов среди бактерий и архей. Эта изменчивость, по-видимому, отражает разнообразие стилей жизни среди соответствующих организмов. Вариации в доле генов, ответственных за передачу сигналов, были количественно отражены в «бактериальном IQ», показателе, который пропорционален квадратному корню от числа белков передачи сигналов (учитывая квадратичное масштабирование) и обратно пропорционален общему количеству генов (Galperin, 2005). IQ отражает способность бактерий и архей отвечать на различные стимулы, приходящие из внешней среды. Соответственно, внутриклеточные симбионты (паразиты) имеют наименьшие значения IQ. Он лишь ненамного выше у организмов с компактными геномами, живущих в стабильной внешней середе, таких как морские цианобактерии, и существенно больше у организмов, живущих в сложной и переменчивой среде, даже у тех, которые обладают сравнительно небольшими геномами.
Горизонтальный перенос генов – определяющий процесс в эволюции прокариот
Повсеместное распространение ГПГ в мире прокариот
Вездесущесть и огромную важность горизонтального переноса генов (ГПГ) в эволюции архей и бактерий можно рассматривать как самую большую новость, выявленную с помощью сравнительного геномного анализа прокариот. Никакое другое открытие не было причиной такого большого количества споров и (порою желчных) дебатов, в которых сталкивались прямо противоположные точки зрения на ГПГ, от утверждений о его повсеместном распространении и всеобъемлющей роли в эволюции бактерий и архей до отрицания любого значимого вклада ГПГ в эволюцию (Gogarten and Townsend, 2005; Kurland et al., 2003; O’Malley and Boucher, 2005). Существование ГПГ, переноса генов между неродственными организмами иным путем, нежели посредством вертикальной передачи реплицированной хромосомы в процессе деления клетки, было осознано задолго до того, как был секвенирован первый геном (Syvanen, 1994). Более того, стало понятно, что ГПГ может происходить исключительно быстро и эффективно – во всяком случае, под давлением отбора, как в случае распространения устойчивости к антибиотикам в популяции патогенных бактерий. Однако, до того как появилась возможность сравнения множества полных геномных последовательностей, ГПГ по молчаливому соглашению рассматривался как маргинальный феномен, возможно важный для таких специфических областей, как эволюция сопротивляемости инфекциям, но по большей части не принимавшийся во внимание при изучении эволюции организмов. Как читатель, вероятно, помнит, сама важность вопроса о роли ГПГ в эволюции была осознана в связи с другим революционным открытием: демонстрацией Вёзе и соавторами того, что филогенетический анализ рРНК прокариот реально возможен и может быть потенциально использован для описания эволюции бактерий и недавно открытых архей. Для большинства биологов трехдоменное эволюционное дерево рРНК, полученное Вёзе, стало синонимом гипотетического древа жизни (ДЖ), исходно постулированного Дарвином, a теперь реально полученного и готового для использования в качестве основы для картирования эволюционных событий всевозможного рода (Pace, 2006). Такова была парадигма, когда сравнительная геномика вызвала революцию, связанную с осознанием роли ГПГ.
Исторически и методологически проблема идентификации актов ГПГ и его влияния на эволюцию бактерий и архей резко различается для случаев (сравнительно) недавних и древних переносов, с одной стороны, и переносов между близкородственными и давно разошедшимися организмами, с другой стороны (Koonin et al., 2001a). Недавние случаи ГПГ, особенно между близкородственными организмами, широко распространены, бесспорны и легко обнаруживаются. Действительно, сравнение геномов бактериальных штаммов предоставляет отчетливые свидетельства большого количества актов ГПГ. Вероятно, наиболее характерным примером является открытие так называемых островов патогенности – генных кластеров, которые несут информацию, типичную для патогенов, подобную той, что содержится в генах, кодирующих различные токсины, компоненты секреторной системы третьего типа и другие подобные системы у бактерий-паразитов, а также похожие «симбиотические острова» у бактерий-симбионтов. Острова патогенности представляют собой протяженные районы генома размером до 100 Кб, которые обычно расположены недалеко от генов тРНК и содержат множество генетического материала профагов, откуда напрашивается предположение, что вставка этих островов в геном была осуществлена при посредничестве бактериофагов (Juhas et al., 2009). Ставший классическим сравнительный геномный анализ энтерогеморрагического штамма O157:H7 и лабораторного штамма K12 бактерии E. coli показал, что патогенный штамм содержит 1,387 дополнительного гена, распределенного между несколькими специфичными для штамма кластерами (островами патогенности), сильно различающимися по размерам. Таким образом, до 30 процентов генов у патогенных штаммов, по-видимому, были приобретены посредством недавнего ГПГ (Perna et al., 2001). Последующий детальный анализ индивидуальных линий O157:H7 показал, что процесс ГПГ непрерывно продолжается, внося свой вклад в различие степени вирулентности этих штаммов (Zhang et al., 2007). Воздействие недавнего ГПГ определенно не ограничивается патогенетическими эффектами. Большинство недавних (случившихся, по оценкам, в течение последних 100 миллионов лет) добавлений в метаболическую сеть E. coli явно были вызваны ГПГ, часто включающим опероны, кодирующие два и более фермента или белка-переносчика одного и того же метаболического пути. Вклад дупликации генов в метаболические инновации оказался в количественном плане существенно менее важным.
Исторически и методологически проблема идентификации актов ГПГ и его влияния на эволюцию бактерий и архей резко различается для случаев (сравнительно) недавних и древних переносов, с одной стороны, и переносов между близкородственными и давно разошедшимися организмами, с другой стороны (Koonin et al., 2001a). Недавние случаи ГПГ, особенно между близкородственными организмами, широко распространены, бесспорны и легко обнаруживаются. Действительно, сравнение геномов бактериальных штаммов предоставляет отчетливые свидетельства большого количества актов ГПГ. Вероятно, наиболее характерным примером является открытие так называемых островов патогенности – генных кластеров, которые несут информацию, типичную для патогенов, подобную той, что содержится в генах, кодирующих различные токсины, компоненты секреторной системы третьего типа и другие подобные системы у бактерий-паразитов, а также похожие «симбиотические острова» у бактерий-симбионтов. Острова патогенности представляют собой протяженные районы генома размером до 100 Кб, которые обычно расположены недалеко от генов тРНК и содержат множество генетического материала профагов, откуда напрашивается предположение, что вставка этих островов в геном была осуществлена при посредничестве бактериофагов (Juhas et al., 2009). Ставший классическим сравнительный геномный анализ энтерогеморрагического штамма O157:H7 и лабораторного штамма K12 бактерии E. coli показал, что патогенный штамм содержит 1,387 дополнительного гена, распределенного между несколькими специфичными для штамма кластерами (островами патогенности), сильно различающимися по размерам. Таким образом, до 30 процентов генов у патогенных штаммов, по-видимому, были приобретены посредством недавнего ГПГ (Perna et al., 2001). Последующий детальный анализ индивидуальных линий O157:H7 показал, что процесс ГПГ непрерывно продолжается, внося свой вклад в различие степени вирулентности этих штаммов (Zhang et al., 2007). Воздействие недавнего ГПГ определенно не ограничивается патогенетическими эффектами. Большинство недавних (случившихся, по оценкам, в течение последних 100 миллионов лет) добавлений в метаболическую сеть E. coli явно были вызваны ГПГ, часто включающим опероны, кодирующие два и более фермента или белка-переносчика одного и того же метаболического пути. Вклад дупликации генов в метаболические инновации оказался в количественном плане существенно менее важным.