В табл. 3–2 описывается несколько примеров ключевых биологических функций, для которых два или несколько неродственных ферментов по-разному представлены в частично дополнительных, но обычно перекрывающихся группах эволюционных линий. Даже эти отдельные примеры показывают, что ЗНОГ происходит в самых различных функциональных системах и путях. В дальнейшем, с заметным увеличением числа секвенированных геномов, стало ясно, что ЗНОГ и утрата генов в отдельных линиях настолько широко распространены, что лишь малое число функций являются действительно мономорфными и вездесущими (то есть представлены ортологичными генами во всех организмах). Вместе с тем универсальное ядро жизни уменьшилось почти до исчезновения: все, что остается универсальным, – это около тридцати генов белков трансляции и три больших субъединицы РНК-полимеразы, а также примерно равное число генов структурных РНК (рРНК и тРНК).
Таблица 3–2. Примеры замещения неортологичных генов.
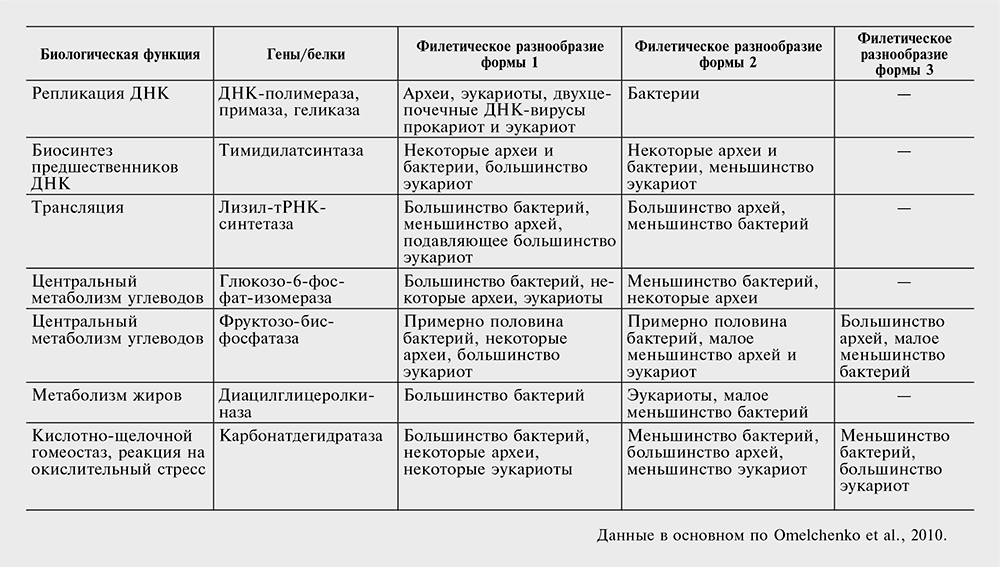
Даже при исключении паразитических бактерий перечень универсальных генов расширяется незначительно (Koonin, 2003). Таким образом, за исключением небольшого числа генов, участвующих в основных этапах передачи информации, не существует универсального генетического ядра жизни, в связи с повсеместными ЗНОГ и потерей генов. Концепция небольшого, универсального набора функций, необходимых для поддержания клетки, остается жизнеспособной, но, учитывая комбинаторику ЗНОГ, этот наименьший набор функциональных ниш может заполняться огромным разнообразием генных ансамблей.
Единицы эволюции и фрактальная структура генетической вселенной
Элементарные события геномной эволюции
Краткий обзор главы
Рекомендуемая дополнительная литература
Глава 4. Геномика, системная биология и универсалии эволюции: эволюция генома как феномен статистической физики
Взаимосвязь между эволюционными и фенотипическими параметрами, универсалии эволюции генов, белков и геномов и физическая модель эволюционного процесса
Таблица 3–2. Примеры замещения неортологичных генов.
Даже при исключении паразитических бактерий перечень универсальных генов расширяется незначительно (Koonin, 2003). Таким образом, за исключением небольшого числа генов, участвующих в основных этапах передачи информации, не существует универсального генетического ядра жизни, в связи с повсеместными ЗНОГ и потерей генов. Концепция небольшого, универсального набора функций, необходимых для поддержания клетки, остается жизнеспособной, но, учитывая комбинаторику ЗНОГ, этот наименьший набор функциональных ниш может заполняться огромным разнообразием генных ансамблей.
Единицы эволюции и фрактальная структура генетической вселенной
Результаты сравнительной геномики приводят к ключевому обобщению, которое позволяет нам выполнять продуктивные эволюционные исследования: основные единицы эволюции могут быть довольно четко определены, и единицы эти – кластеры ортологичных генов, или эволюционные домены (КОГ), или, еще точнее, линии эволюционирующих ортологичных генов (доменов). Истории отдельных генов часто сложны (а во многих случаях даже чрезвычайно сложны) и включают в себя множественные утраты генов, дупликацию и горизонтальный перенос (ниже в настоящей книге мы обсудим эти явления подробнее, см. гл. 5 и 7). Предрасположенность генов к дупликации, утрате и переносу варьирует в широких пределах. Однако, невзирая на все эти осложнения, атомарное свойство наборов ортологичных генов твердо соблюдается: КОГ суть естественные элементы генетической вселенной.
Генетическая (геномная) вселенная (это только метафора, но удобная и, возможно, продуктивная) может быть представлена как развивающееся пространство-время, заполненное кластерами, состоящими из генов (то есть КОГ), или, точнее, эволюционирующими линиями ортологов, элементарными единицами эволюции. Ортология легче всего прослеживается между прокариотическими генами, так что здесь мы обсудим прокариотическую область геномной вселенной. Тенденции среди эукариотов в принципе похожи, но осложнены распространенной мультидоменной организацией белков и обширной паралогией. В нашем геномном пространстве заметно характерное распределение КОГ по геномам, хорошо аппроксимируемое тремя экспонентами с разными показателями, которые делят генную популяцию на три класса (см. рис. 3-13, а – в; Koonin and Wolf, 2008b).
1. (Почти) универсальные гены, те, что представлены в (почти) всех геномах клеточных форм жизни, составляют лишь малую часть генетической вселенной: это ядро клеточной жизни состоит, самое большее, из приблизительно 70 генов. В каждом конкретном геноме доля этих «ядерных» генов составляет не более 10 процентов, если говорить о самых маленьких геномах клеточных форм жизни (паразитических бактерий, таких как M. genitalium), но обычно ближе к 1 проценту или менее от общего числа генов (см. рис. 3-14).
2. Умеренно консервативная генная оболочка состоит из КОГ, представленных в самых разнообразных геномах, но не в подавляющем их большинстве. Недавний анализ имеющихся прокариотических геномов дает число КОГ оболочки около 5000. Гены оболочки составляют большую часть числа генов в любом геноме (см. рис. 3-14).
3. Малоконсервативное «облако» состоит из КОГ, встречающихся в узких группах организмов, и «генов-сирот» – генов в открытых рамках считывания (ОРС), обнаруженных пока что в одном-единственном геноме, но гомологи которых обычно обнаруживаются во вновь появляющихся геномных данных. Гены «облака» составляют переменную долю в каждом геноме, обычно в интервале 10–30 процентов от общего числа генов (см. рис. 3-14).
Примечательно, что эта структура является самоподобной, или фрактальной: те же три компонента – крошечное ядро, сравнительно большая оболочка и огромное «облако» – проявляются на любом уровне, где бы ни рассекалось генное пространство-время, от всего мира прокариот и до узких групп бактерий (см. рис. 3-14). Мы возвратимся к последствиям этой фрактальности геномного пространства-времени прокариот в главе 5. Заметим, однако, что эволюционная модель, которая объясняла бы наблюдаемую фрактальность, еще ожидает своей разработки[36].
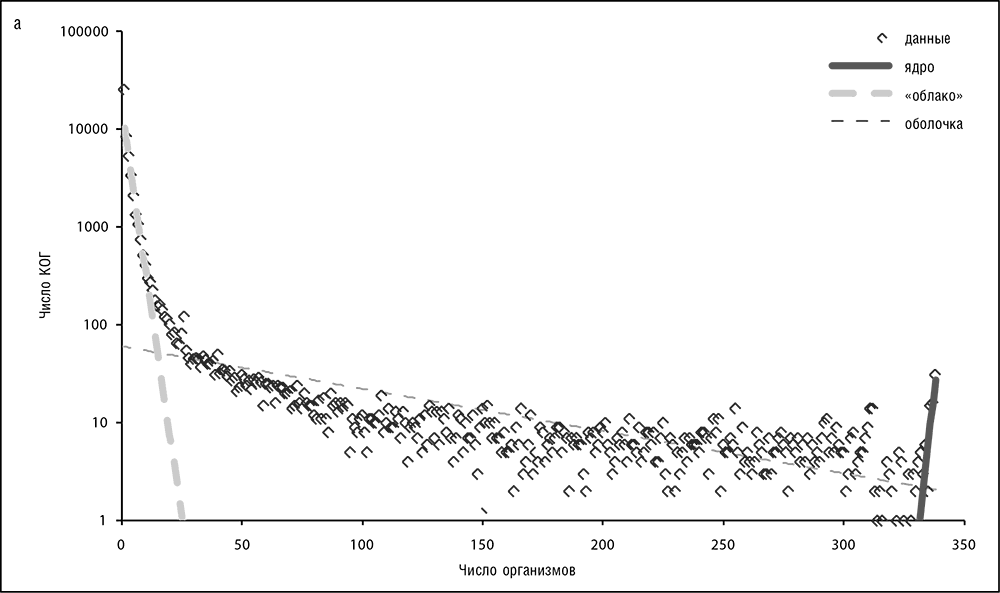 Рис. 3-13 а. Глубокий уровень: 338 прокариот из базы данных EggNOG (Jensen et al., 2008)
Рис. 3-13 а. Глубокий уровень: 338 прокариот из базы данных EggNOG (Jensen et al., 2008)
 Рис. 3-13 б. Средний уровень: 41 архей из базы данных arCOG (Makarova et al., 2007b)
Рис. 3-13 б. Средний уровень: 41 архей из базы данных arCOG (Makarova et al., 2007b)
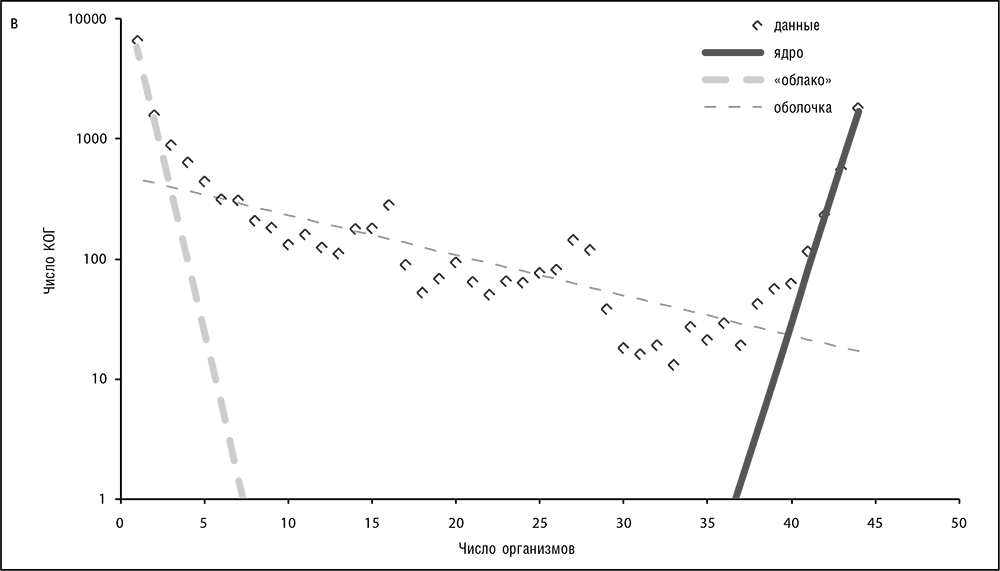 Рис. 3-13 в. Мелкий уровень: 44 вида Escherichia, Shigella и Salmonella из базы данных COG (Tatusov et al., 2003). Регрессия данных экспоненциальными функциями на всех трех рисунках (Koonin and Wolf, 2008b) изображена пунктирными и непрерывными линиями.
Рис. 3-13 в. Мелкий уровень: 44 вида Escherichia, Shigella и Salmonella из базы данных COG (Tatusov et al., 2003). Регрессия данных экспоненциальными функциями на всех трех рисунках (Koonin and Wolf, 2008b) изображена пунктирными и непрерывными линиями.
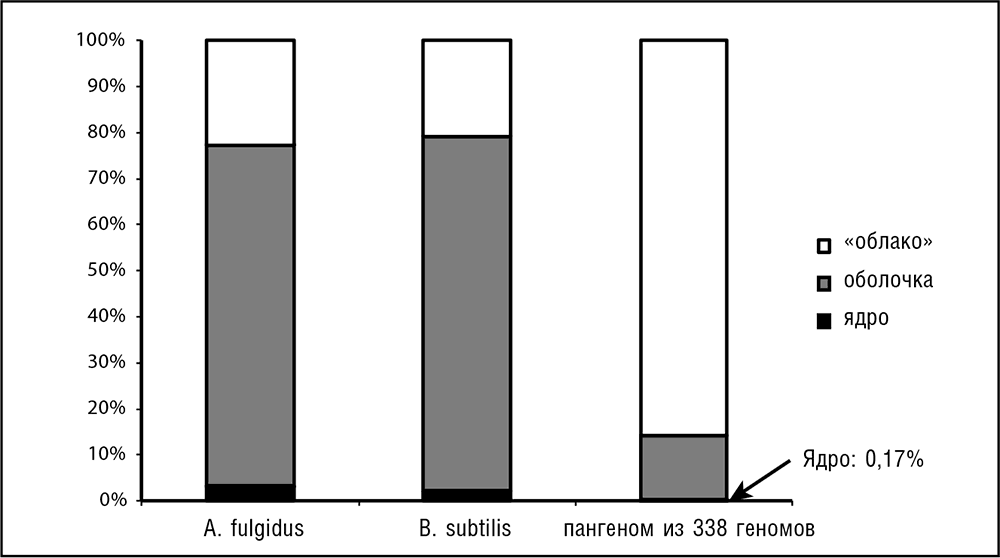 Рис. 3-14. Вклад ядра, оболочки и «облака» в состав индивидуальных геномов и генной вселенной как целого. Расчет произведен по данным из базы EggNOG (Jensen et al., 2008). A. fulgidus – архея Archaeoglobus fulgidus; B. subtilis – бактерия Bacillus subtilis.
Рис. 3-14. Вклад ядра, оболочки и «облака» в состав индивидуальных геномов и генной вселенной как целого. Расчет произведен по данным из базы EggNOG (Jensen et al., 2008). A. fulgidus – архея Archaeoglobus fulgidus; B. subtilis – бактерия Bacillus subtilis.
Следует обратить внимание на кажущийся парадокс в распределении КОГ в генном пространстве. Хотя в каждом геноме большинство генов относятся к оболочке, то есть являются общими с дальнородственными организмами, при рассмотрении всей генной вселенной оказывается, что гены (или, вернее, КОГ) ядра и оболочки составляют лишь незначительное меньшинство (см. рис. 3-14). Вполне очевидно, что эта разница возникает потому, что КОГ оболочки представлены во многих геномах, в то время как КОГ «облака», особенно «гены-сироты», являются редкими или уникальными. С учетом этой характерной структуры вселенной генов эволюционные реконструкции неизбежно приводят к картине динамичной эволюции генома, где многочисленные гены (в основном из «облака» и, в меньшей степени, из оболочки) утрачиваются, а многие другие приобретаются путем ГПГ (в основном у прокариот), а также в результате многочисленных дупликаций, в первую очередь у эукариот (см. ниже в этой главе).
Генетическая (геномная) вселенная (это только метафора, но удобная и, возможно, продуктивная) может быть представлена как развивающееся пространство-время, заполненное кластерами, состоящими из генов (то есть КОГ), или, точнее, эволюционирующими линиями ортологов, элементарными единицами эволюции. Ортология легче всего прослеживается между прокариотическими генами, так что здесь мы обсудим прокариотическую область геномной вселенной. Тенденции среди эукариотов в принципе похожи, но осложнены распространенной мультидоменной организацией белков и обширной паралогией. В нашем геномном пространстве заметно характерное распределение КОГ по геномам, хорошо аппроксимируемое тремя экспонентами с разными показателями, которые делят генную популяцию на три класса (см. рис. 3-13, а – в; Koonin and Wolf, 2008b).
1. (Почти) универсальные гены, те, что представлены в (почти) всех геномах клеточных форм жизни, составляют лишь малую часть генетической вселенной: это ядро клеточной жизни состоит, самое большее, из приблизительно 70 генов. В каждом конкретном геноме доля этих «ядерных» генов составляет не более 10 процентов, если говорить о самых маленьких геномах клеточных форм жизни (паразитических бактерий, таких как M. genitalium), но обычно ближе к 1 проценту или менее от общего числа генов (см. рис. 3-14).
2. Умеренно консервативная генная оболочка состоит из КОГ, представленных в самых разнообразных геномах, но не в подавляющем их большинстве. Недавний анализ имеющихся прокариотических геномов дает число КОГ оболочки около 5000. Гены оболочки составляют большую часть числа генов в любом геноме (см. рис. 3-14).
3. Малоконсервативное «облако» состоит из КОГ, встречающихся в узких группах организмов, и «генов-сирот» – генов в открытых рамках считывания (ОРС), обнаруженных пока что в одном-единственном геноме, но гомологи которых обычно обнаруживаются во вновь появляющихся геномных данных. Гены «облака» составляют переменную долю в каждом геноме, обычно в интервале 10–30 процентов от общего числа генов (см. рис. 3-14).
Примечательно, что эта структура является самоподобной, или фрактальной: те же три компонента – крошечное ядро, сравнительно большая оболочка и огромное «облако» – проявляются на любом уровне, где бы ни рассекалось генное пространство-время, от всего мира прокариот и до узких групп бактерий (см. рис. 3-14). Мы возвратимся к последствиям этой фрактальности геномного пространства-времени прокариот в главе 5. Заметим, однако, что эволюционная модель, которая объясняла бы наблюдаемую фрактальность, еще ожидает своей разработки[36].
Следует обратить внимание на кажущийся парадокс в распределении КОГ в генном пространстве. Хотя в каждом геноме большинство генов относятся к оболочке, то есть являются общими с дальнородственными организмами, при рассмотрении всей генной вселенной оказывается, что гены (или, вернее, КОГ) ядра и оболочки составляют лишь незначительное меньшинство (см. рис. 3-14). Вполне очевидно, что эта разница возникает потому, что КОГ оболочки представлены во многих геномах, в то время как КОГ «облака», особенно «гены-сироты», являются редкими или уникальными. С учетом этой характерной структуры вселенной генов эволюционные реконструкции неизбежно приводят к картине динамичной эволюции генома, где многочисленные гены (в основном из «облака» и, в меньшей степени, из оболочки) утрачиваются, а многие другие приобретаются путем ГПГ (в основном у прокариот), а также в результате многочисленных дупликаций, в первую очередь у эукариот (см. ниже в этой главе).
Элементарные события геномной эволюции
Теперь, определив единицы геномной эволюции и разработав идею организации вселенной генов, мы можем осмысленно дополнить эти понятия списком основных операций, элементарных событий эволюции генома, которые можно будет сравнить с элементарными событиями эволюции отдельных генов. Алфавиты элементарных событий довольно кратки и фактически подобны (изоморфны) на соответствующих уровнях (см. табл. 3–3). Однако относительный вклад и частота различных типов событий разнятся в эволюции генов и геномов самым коренным образом. Существенное различие между эволюцией отдельных генов и целых геномов заключается в особой важности и высокой частоте дупликации генов, в отличие от много более ограниченного вклада внутригенных дупликаций. Далее, внутригенные рекомбинации редко закрепляются в эволюции, за исключением близких геномов, a важнейшие механизмы перестройки генома, такие как инверсии и транслокации, не играют особой роли в эволюции отдельных генов. В итоге различия в относительном вкладе разнообразных элементарных механизмов (см. табл. 3–2) лежат в основе значительно более динамичного характера эволюции геномов по сравнению с эволюцией отдельных генов.
Таблица 3–3. Сравнение элементарных событий эволюции гена и генома.
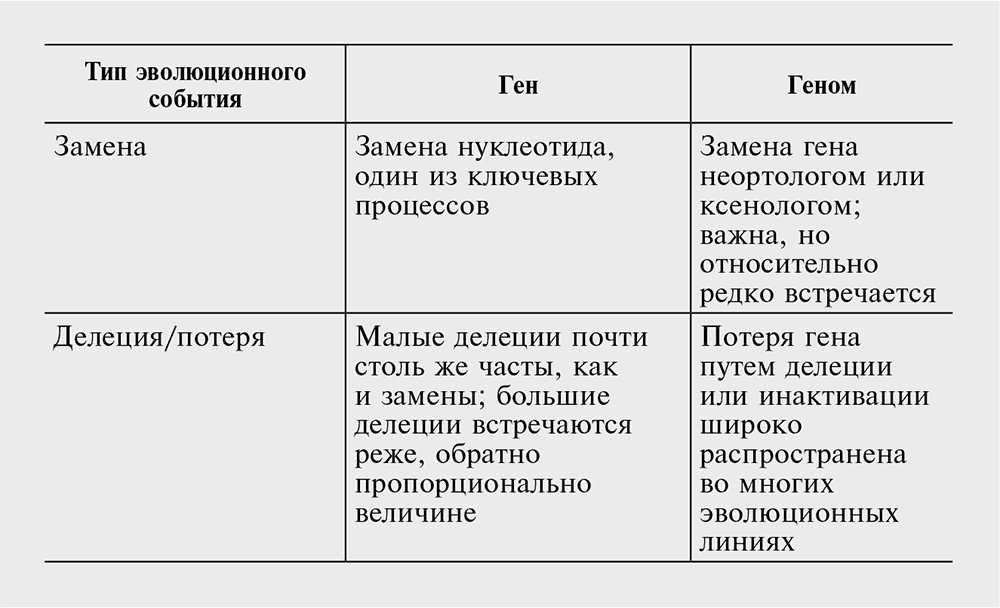
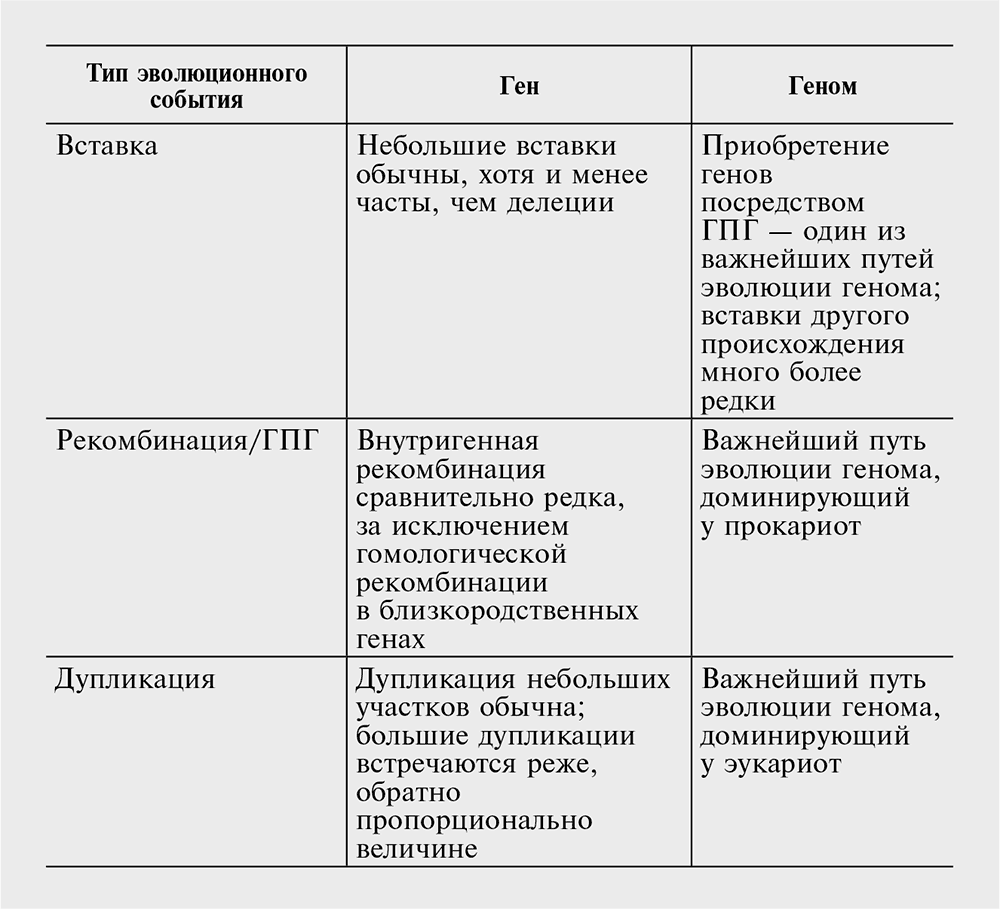
Таблица 3–3. Сравнение элементарных событий эволюции гена и генома.
Краткий обзор главы
Сравнительная геномика раскрывает примечательный контраст между относительной эволюционной устойчивостью отдельных генов, многие из которых сохраняют значительное сходство на протяжении сотен миллионов или даже миллиардов лет эволюции, и пластичностью состава и архитектуры генома, которые изменяются на несколько порядков быстрее. Отсюда возникает характерное устройство вселенной генов, в котором сравнительно небольшое число плотных кластеров образуют ядро, гены которого представлены в большинстве геномов, в то время как большую часть пространства-времени занимает огромное количество все более разреженных «туманностей», состоящих из редких генов. Поразительно, что организация генетической вселенной явственно фрактальна, то есть проявляется на всех масштабах эволюционных расстояний.
Атомарная сущность генов (или, точнее, КОГ, ортологичных эволюционных линий) лежит в основе всей исследовательской программы сравнительной геномики: сравнение геномов оказывается весьма информативным, несмотря на нетривиальные отношения между отдельными генами и геномами, обусловленные изменчивостью геномной архитектуры.
Геномные ландшафты различных форм жизни – распределение ограничений по геномным сайтам – разнообразны и сложны. Компактные геномы вирусов, прокариот и, в меньшей степени, одноклеточных эукариот в основном занимают «высокогорные плато», так что почти все сайты подвергаются существенным ограничениям. Геномные ландшафты многоклеточных эукариот состоят в основном из «долин» со слабыми ограничениями, разделенных редкими «гребнями» сильного отбора. Эти отличия отражают разные эволюционные режимы, которые мы обсудим в главе 8. Парадоксально, но именно «неэффективность» режима эволюции, характерного для многоклеточных эукариот, позволяет организационной сложности возникнуть. Этот парадокс должен заставить задуматься всех неравнодушных к идее эволюционного «прогресса». Мы вернемся к подробному обсуждению этого вопроса в главах 8 и 13.
Атомарная сущность генов (или, точнее, КОГ, ортологичных эволюционных линий) лежит в основе всей исследовательской программы сравнительной геномики: сравнение геномов оказывается весьма информативным, несмотря на нетривиальные отношения между отдельными генами и геномами, обусловленные изменчивостью геномной архитектуры.
Геномные ландшафты различных форм жизни – распределение ограничений по геномным сайтам – разнообразны и сложны. Компактные геномы вирусов, прокариот и, в меньшей степени, одноклеточных эукариот в основном занимают «высокогорные плато», так что почти все сайты подвергаются существенным ограничениям. Геномные ландшафты многоклеточных эукариот состоят в основном из «долин» со слабыми ограничениями, разделенных редкими «гребнями» сильного отбора. Эти отличия отражают разные эволюционные режимы, которые мы обсудим в главе 8. Парадоксально, но именно «неэффективность» режима эволюции, характерного для многоклеточных эукариот, позволяет организационной сложности возникнуть. Этот парадокс должен заставить задуматься всех неравнодушных к идее эволюционного «прогресса». Мы вернемся к подробному обсуждению этого вопроса в главах 8 и 13.
Рекомендуемая дополнительная литература
Ellegren, H. (2008) Comparative Genomics and the Study of Evolution by Natural Selection. Molecular Ecology 17: 4,586—4,596.
Обзор факторов отбора, важных для эволюции разных классов геномных последовательностей.
Koonin, E. V. (2005) Orthologs, Paralogs, and Evolutionary Genomics. Annual Review of Genetics 39: 309–338.
Детальное обсуждение концепций ортологии и паралогии, а также определенных категорий эволюционных отношений между генами в этих широких классах.
Koonin, E. V. (2009) Evolution of Genome Architecture. International Journal of Biochemistry and Cell Biology 41: 298–306.
Обзор разнообразия и эволюционных тенденций геномных архитектур различных форм клеточной жизни.
Koonin, E. V. (2003) Comparative Genomics, Minimal Gene-Sets, and the Last Universal Common Ancestor. Nature Reviews Microbiology 1: 127–136.
Критическое обсуждение концепции минимального набора генов и ее применения к организмам, имеющим различный стиль жизни, а также сравнение минимальных наборов и реконструкция предковых геномов.
Koonin, E. V., and Y. I. Wolf. (2010) Constraints and Plasticity in Genome and Molecular-Phenome Evolution. Nature Reviews Genetics 11: 487–498.
Попытка исчерпывающей полногеномной переписи эволюционных ограничений, действующих на различные классы последовательностей и сайтов в геномах.
Koonin, E. V., and Y. I. Wolf. (2008) Genomics of Bacteria and Archaea: The Emerging Dynamic View of the Prokaryotic World. Nucleic Acids Research 36: 6,688—6,719.
Детальный обзор геномики прокариот с особым упором на динамику генома, в том числе ГПГ.
Levitt, M. Nature of the Protein Universe. (2009) Proceedings of the National Academy of Sciences USA 106: 11,079—11,084.
Подробное исследование происхождения новизны в эволюции белка. Делается вывод о том, что ключевым механизмом порождения новизны явилось возникновение мультидоменной архитектуры.
Lynch, Michael. (2007) The Origins of Genome Architecture. Sunderland, MA: Sinauer Associates.
Принципиально важная книга по неадаптивной теории эволюции геномной сложности и ее различным последствиям (подробное обсуждение в гл. 8).
Wilkins, A. S. (1997) Canalization: A Molecular Genetic Perspective. Bioessays 19: 257–262.
Переоценка Уоддингтоновой концепции канализации в контексте современной эволюционной биологии.
Обзор факторов отбора, важных для эволюции разных классов геномных последовательностей.
Koonin, E. V. (2005) Orthologs, Paralogs, and Evolutionary Genomics. Annual Review of Genetics 39: 309–338.
Детальное обсуждение концепций ортологии и паралогии, а также определенных категорий эволюционных отношений между генами в этих широких классах.
Koonin, E. V. (2009) Evolution of Genome Architecture. International Journal of Biochemistry and Cell Biology 41: 298–306.
Обзор разнообразия и эволюционных тенденций геномных архитектур различных форм клеточной жизни.
Koonin, E. V. (2003) Comparative Genomics, Minimal Gene-Sets, and the Last Universal Common Ancestor. Nature Reviews Microbiology 1: 127–136.
Критическое обсуждение концепции минимального набора генов и ее применения к организмам, имеющим различный стиль жизни, а также сравнение минимальных наборов и реконструкция предковых геномов.
Koonin, E. V., and Y. I. Wolf. (2010) Constraints and Plasticity in Genome and Molecular-Phenome Evolution. Nature Reviews Genetics 11: 487–498.
Попытка исчерпывающей полногеномной переписи эволюционных ограничений, действующих на различные классы последовательностей и сайтов в геномах.
Koonin, E. V., and Y. I. Wolf. (2008) Genomics of Bacteria and Archaea: The Emerging Dynamic View of the Prokaryotic World. Nucleic Acids Research 36: 6,688—6,719.
Детальный обзор геномики прокариот с особым упором на динамику генома, в том числе ГПГ.
Levitt, M. Nature of the Protein Universe. (2009) Proceedings of the National Academy of Sciences USA 106: 11,079—11,084.
Подробное исследование происхождения новизны в эволюции белка. Делается вывод о том, что ключевым механизмом порождения новизны явилось возникновение мультидоменной архитектуры.
Lynch, Michael. (2007) The Origins of Genome Architecture. Sunderland, MA: Sinauer Associates.
Принципиально важная книга по неадаптивной теории эволюции геномной сложности и ее различным последствиям (подробное обсуждение в гл. 8).
Wilkins, A. S. (1997) Canalization: A Molecular Genetic Perspective. Bioessays 19: 257–262.
Переоценка Уоддингтоновой концепции канализации в контексте современной эволюционной биологии.
Глава 4. Геномика, системная биология и универсалии эволюции: эволюция генома как феномен статистической физики
В предыдущей главе была подчеркнута относительная стабильность отдельных генов, составляющая яркий контраст динамизму геномной эволюции. Если гены или домены принять за атомарные единицы геномной эволюции, тогда геномы можно рассмотреть как статистические ансамбли таких единиц. Мы можем продолжить эту очень упрощенную, но очевидно не бессмысленную и потенциально продуктивную физическую аналогию и рассмотреть геномы как структуры, подобные газу или жидкости, в которых силы межмолекулярного взаимодействия хоть и являются важными параметрами, но слабы по сравнению с внутримолекулярными взаимодействиями (лежащими в основе стабильности молекул), в отличие от твердых тел, в которых межмолекулярные взаимодействия сильны и имеют определяющее значение.
Из статистической физики известно, что поведение ансамбля слабовзаимодействующих частиц (молекул) следует простым и универсальным статистическим закономерностям, таким как распределение Больцмана для скоростей частиц. Аналогия между ансамблями генов (геномами) и ансамблями молекул (газами и жидкостями) наталкивает нас на поиск статистических закономерностей в функционировании и эволюции генома. Более того, размышляя таким образом, мы можем с некоторой степенью уверенности предположить, что эти статистические закономерности должны представлять собой математически простые, универсальные законы распределения значений определенных параметров, описывающих процесс эволюции. Мы убедимся в этой главе, что поиск таких эволюционных универсалий – дело далеко не безнадежное.
Перед обсуждением статистических свойств генных ансамблей необходимо обратить внимание на еще одно ведущее направление биологических исследований первой декады третьего тысячелетия, представляющее собой новую область науки, часто называемую, может быть не очень удачно, системной биологией. Системная биология провозглашает своей конечной целью построение моделей и понимание функционирования биологических систем во всей их сложности. Реальное положение дел на данном этапе становления этой области исследований заключается в том, что основное внимание направлено на агрегацию обширных данных специфического типа, таких как транскриптомы (совокупность всех экспрессируемых РНК клетки, ткани или организма), протеомы (совокупность всех экспрессированных белков), метаболомы (совокупность всех метаболитов) и другие «-омы» (Bruggeman and Westerhoff, 2007; Koonin and Wolf, 2008a). Все эти «-омы» описываются системной биологией с помощью количественных понятий, таких как концентрация белка или метаболита.
Так же как и генетику в ее первые годы, системную биологию многие ученые приняли за скучную «большую науку» и слишком хлопотливое занятие. (Подозреваю, что это отношение до сих пор преобладает.) Так же как и с генетикой, первый взгляд оказался, мягко говоря, недальновидным. Наличие высококачественных данных по генной экспрессии, генетическим и белок-белковым взаимодействиям, локализации белка в клетке и других данных системного уровня в масштабе генома открыло новые измерения эволюционного анализа (иначе иногда называемого эволюционной системной биологией) и обеспечило его взаимопроникновение с эволюционной геномикой. В исследованиях системной биологии, в масштабах генома, уже были открыты нетривиальные связи между эволюцией генных последовательностей, генной экспрессией, структурой белка и другими характеристиками генов и белков. Эти открытия в целом оказались совместимыми с точкой зрения на геном как на статистический ансамбль генов и дали возможность в новом свете рассмотреть селективную и нейтральную составляющие эволюции структуры и функций генома.
Из статистической физики известно, что поведение ансамбля слабовзаимодействующих частиц (молекул) следует простым и универсальным статистическим закономерностям, таким как распределение Больцмана для скоростей частиц. Аналогия между ансамблями генов (геномами) и ансамблями молекул (газами и жидкостями) наталкивает нас на поиск статистических закономерностей в функционировании и эволюции генома. Более того, размышляя таким образом, мы можем с некоторой степенью уверенности предположить, что эти статистические закономерности должны представлять собой математически простые, универсальные законы распределения значений определенных параметров, описывающих процесс эволюции. Мы убедимся в этой главе, что поиск таких эволюционных универсалий – дело далеко не безнадежное.
Перед обсуждением статистических свойств генных ансамблей необходимо обратить внимание на еще одно ведущее направление биологических исследований первой декады третьего тысячелетия, представляющее собой новую область науки, часто называемую, может быть не очень удачно, системной биологией. Системная биология провозглашает своей конечной целью построение моделей и понимание функционирования биологических систем во всей их сложности. Реальное положение дел на данном этапе становления этой области исследований заключается в том, что основное внимание направлено на агрегацию обширных данных специфического типа, таких как транскриптомы (совокупность всех экспрессируемых РНК клетки, ткани или организма), протеомы (совокупность всех экспрессированных белков), метаболомы (совокупность всех метаболитов) и другие «-омы» (Bruggeman and Westerhoff, 2007; Koonin and Wolf, 2008a). Все эти «-омы» описываются системной биологией с помощью количественных понятий, таких как концентрация белка или метаболита.
Так же как и генетику в ее первые годы, системную биологию многие ученые приняли за скучную «большую науку» и слишком хлопотливое занятие. (Подозреваю, что это отношение до сих пор преобладает.) Так же как и с генетикой, первый взгляд оказался, мягко говоря, недальновидным. Наличие высококачественных данных по генной экспрессии, генетическим и белок-белковым взаимодействиям, локализации белка в клетке и других данных системного уровня в масштабе генома открыло новые измерения эволюционного анализа (иначе иногда называемого эволюционной системной биологией) и обеспечило его взаимопроникновение с эволюционной геномикой. В исследованиях системной биологии, в масштабах генома, уже были открыты нетривиальные связи между эволюцией генных последовательностей, генной экспрессией, структурой белка и другими характеристиками генов и белков. Эти открытия в целом оказались совместимыми с точкой зрения на геном как на статистический ансамбль генов и дали возможность в новом свете рассмотреть селективную и нейтральную составляющие эволюции структуры и функций генома.
Взаимосвязь между эволюционными и фенотипическими параметрами, универсалии эволюции генов, белков и геномов и физическая модель эволюционного процесса
В предыдущей главе было показано, что белок-кодирующие гены (по крайней мере в отношении мутационных замен, приводящих к изменению аминокислот в кодируемом белке) принадлежат к числу наиболее консервативных последовательностей генома. Однако уже на раннем этапе исследований в молекулярной эволюции стало понятно, что скорости эволюционирования белок-кодирующих генов могут очень сильно разниться (Wilson et al., 1977). Этот широкий разброс значений в общем объясняли существованием широкого спектра функций белка, которые по-разному ограничивают скорость эволюции соответствующих генов. В самом деле, кажется само собой разумеющимся, что огромная роль ДНК-полимеразы, сложнейшего фермента, который катализирует встраивание комплементарных матрице нуклеотидов в растущую цепь ДНК, требует значительного ограничения на скорость эволюции для соответствующей ей генной последовательности, в то время как, например, для структурного белка, чья единственная задача состоит в поддержании целостности ядерного матрикса, такого сильного ограничения не требуется. Фундаментальное представление о том, что эволюция белок-кодирующих генов может сводиться не только к уникальным особенностям молекулярной структуры и функции белков, возникло уже на этом раннем этапе. В богатой идеями обзорной статье, опубликованной Аланом Вильсоном и коллегами в 1977 году, выдвигается гипотеза о том, что скорость эволюции генных последовательностей зависит как от уникальных функций кодируемого белка, так и от важности этого белка для выживания организма (Wilson et al., 1977). Однако в то время не было прямых способов изучения эволюционных ограничений, так что эти идеи, хоть и интригующие, тогда находились всецело в области умозрительных построений.
В начале третьего тысячелетия геномика и системная биология полностью преобразили область эволюционных исследований. Доступность множества данных по геномным последовательностям позволила проанализировать и сравнить распределения скоростей эволюции для полных наборов ортологичных генов в различных таксонах, а также изучить взаимосвязи скоростей эволюции ортологов в различных эволюционных линиях. Значения скоростей эволюции по несинонимичным сайтам в ортологичных генах могут различаться на три-четыре порядка, и это распределение значений гораздо шире, чем распределение скоростей по синонимичным сайтам. Замечательно, что формы графиков распределений по ортологичным белкам исключительно похожи, практически одинаковы для всех изученных клеточных форм жизни, от бактерий и архей до млекопитающих (см. рис. 4–2; Grishin et al., 2000; Wolf et al., 2009). Все эти распределения имеют так называемую логарифмически нормальную форму, то есть распределение логарифма эволюционной скорости близко к нормальному (распределению Гаусса, функция плотности вероятности которого имеет колоколообразную форму). В теории случайных процессов такая форма обычно представляет собой результат произведения многих независимых случайных величин. Универсальность функции распределения среди различных организмов, обладающих глубокими различиями в функциональной организации и сильно различающихся по размеру геномов, представляется неожиданной и может указывать на существование фундаментальных, простых объяснений, которые мы и обсудим в этой главе.
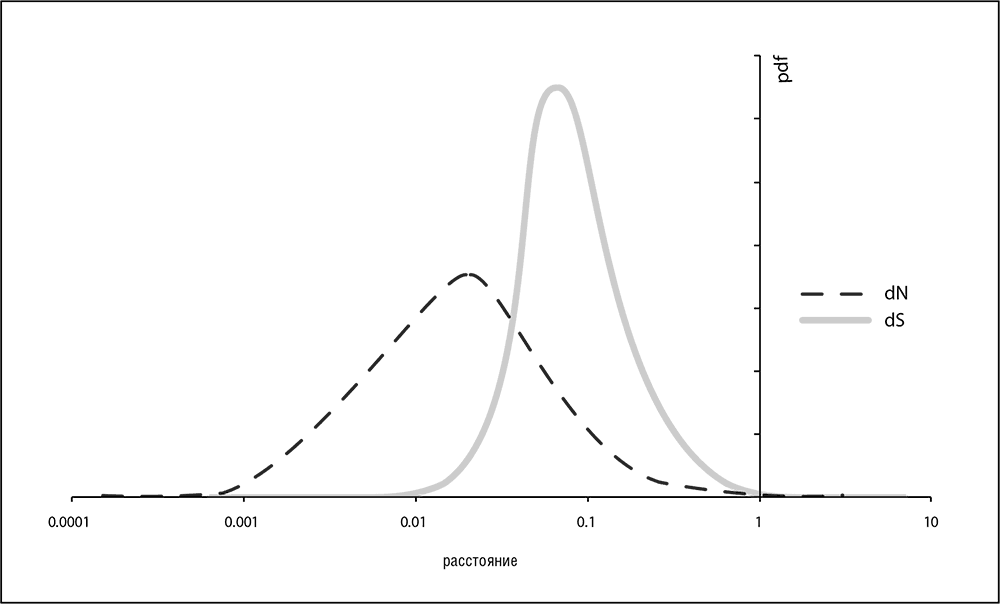 Рис. 4–1. Распределения скорости эволюции по несинонимичным и синонимичным сайтам в ортологичных генах человека и мыши: dN = скорость эволюции по несинонимичным сайтам; dS = по синонимичным; pdf = функция плотности вероятности. Данные из Wolf et al., 2009; для расчетов использовался пакет PALM (Yang, 2007)
Рис. 4–1. Распределения скорости эволюции по несинонимичным и синонимичным сайтам в ортологичных генах человека и мыши: dN = скорость эволюции по несинонимичным сайтам; dS = по синонимичным; pdf = функция плотности вероятности. Данные из Wolf et al., 2009; для расчетов использовался пакет PALM (Yang, 2007)
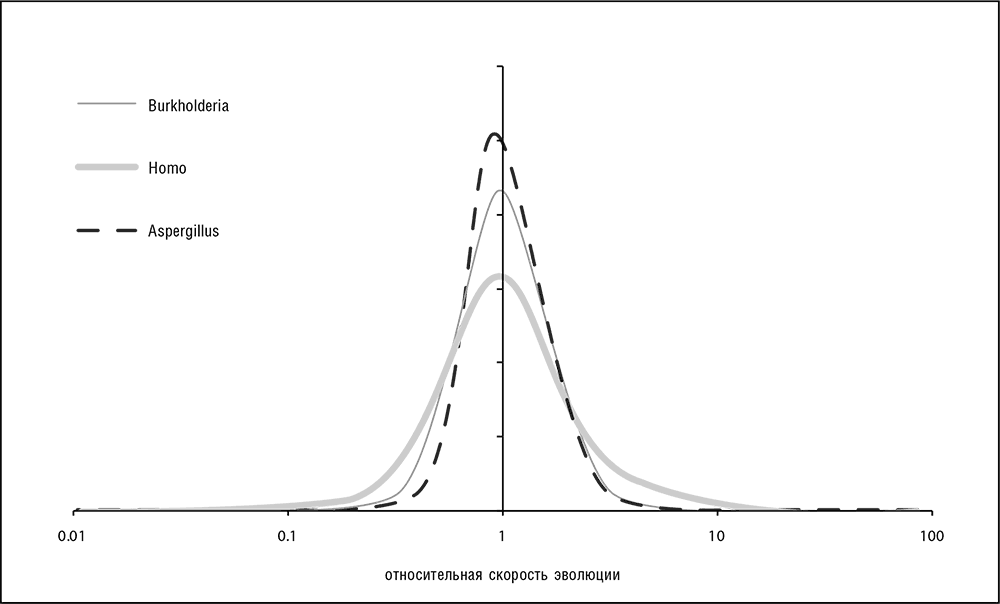 Рис. 4–2. Распределения скорости эволюции в наборах ортологичных генов бактерий и эукариот; Burkholderia = распределения для ортологов Burkholderia cenocepacia и Burkholderia vietnamiensis (протеобактерия); Homo = для ортологов человека и макаки-резус (приматы). Aspergillus = для ортологов Aspergillus fumigatus и Neosartorya fischeri (аскомицеты). Данные из Lobkovsky et al., 2010; для расчетов использовался пакет PALM (Yang, 2007)
Рис. 4–2. Распределения скорости эволюции в наборах ортологичных генов бактерий и эукариот; Burkholderia = распределения для ортологов Burkholderia cenocepacia и Burkholderia vietnamiensis (протеобактерия); Homo = для ортологов человека и макаки-резус (приматы). Aspergillus = для ортологов Aspergillus fumigatus и Neosartorya fischeri (аскомицеты). Данные из Lobkovsky et al., 2010; для расчетов использовался пакет PALM (Yang, 2007)
Прогресс в системной биологии позволил измерить корреляции между скоростью эволюции и всеми возможными молекулярно-фенотипическими величинами, такими как уровень экспрессии, концентрации белков, белок-белковые взаимодействия, фенотипический эффект генной мутации и другими (Koonin and Wolf, 2006). Эти поиски корреляций стали практически самостоятельной областью исследований, цель которых, однако, состоит не в описании самих корреляций, а в построении физически осмысленной модели эволюции геномов и феномов. Было найдено много важных корреляций, что позволило увидеть существование некоторых закономерностей, несмотря на «зашумленность» молекулярно-фенотипических данных (особенно данных, полученных на ранних этапах исследований). На рис. 4–3 представлена простая и наглядная, хоть и неизбежно упрощенная общая картина результатов исследований (Wolf et al., 2006). Обобщение результатов показывает, что существуют два обширных класса переменных:
1. Интенсивные, эволюционные переменные – различные скорости геномных изменений, включая эволюцию последовательностей, потерю гена, перестройку генома и другие виды эволюционных процессов.
2. Экстенсивные, фенотипические переменные – скорость экспрессии, скорость трансляции, концентрация белка, частота взаимодействия с другими изучаемыми объектами.
Корреляции внутри каждого из двух классов обычно положительные, а корреляции между двумя классами – отрицательные (рис. 4–3). Эта закономерность предполагает модель «статуса генов», в которой высокостатусные гены эволюционируют медленно, имеют высокий уровень экспрессии и взаимодействуют со многими другими генами. Гены с низким статусом меняются быстро и имеют низкий уровень экспрессии и меньшее число партнеров (рис. 4–4).
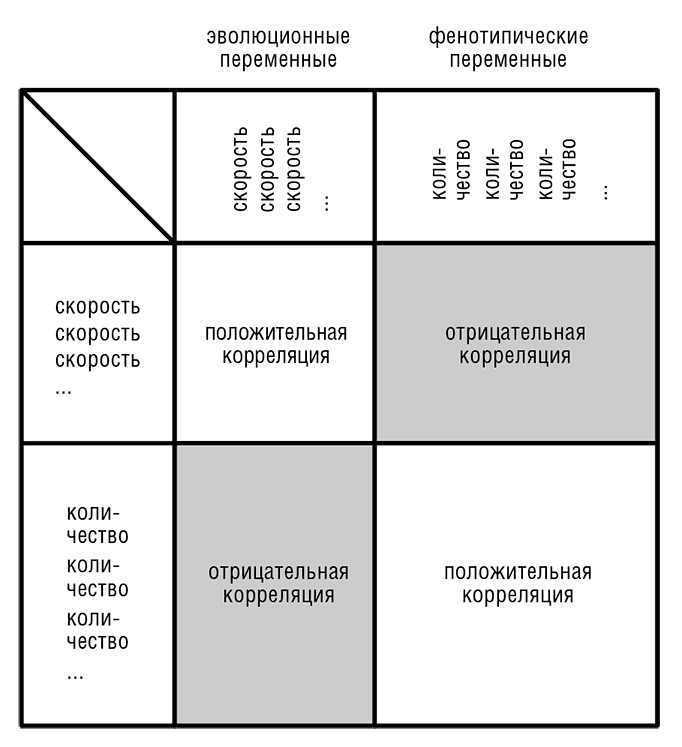 Рис. 4–3. Схематическая обобщенная картина корреляций эволюционных и молекулярно-фенотипических переменных.
Рис. 4–3. Схематическая обобщенная картина корреляций эволюционных и молекулярно-фенотипических переменных.
Сильнейшая, универсальная связь между эволюционными и молекулярно-фенотипическими переменными состоит в отрицательной корреляции скорости эволюции белок-кодирующих генов и уровня экспрессии: высокоэкспрессированные гены эволюционируют медленно. Эта зависимость наблюдается у всех организмов, для которых есть данные по экспрессии генов (Drummond et al., 2006; Drummond and Wilke, 2008; Pal et al., 2001). Поскольку, как отмечено выше, существует положительная корреляция между Ка и Ks, неудивительно, что скорости эволюции синонимических и несинонимических сайтов связаны с уровнем экспрессии гена качественно одним и тем же образом. Более неожиданно то, что зависимость между экспрессией и скоростью эволюции соблюдается и для 3’-нетранслируемого участка (НТУ), хотя и не обнаружена для 5’-НТУ (Jordan et al., 2004). Эта универсальная отрицательная корреляция проявляется еще сильнее, если сравнивать скорость эволюции напрямую с экспериментально измеренными концентрациями белка (Schrimpf et al., 2009).
Открытие универсальной связи между экспрессией генов и их эволюцией стимулировало смелую попытку новой интерпретации, согласно которой эволюция белков определяется в большей степени принципами структуры и укладки белка, общими для всех организмов, чем его уникальными биологическими функциями. Было выдвинуто предположение, впервые – в работе Алана Драмонда и Клауса Вилке, о том, что главным фактором отбора в эволюции белка является его устойчивость к неправильной укладке. Согласно этой гипотезе, влияние мутации, как геномной, так и фенотипической (вызванной ошибками трансляции), на приспособленность организма в первую очередь рассматривается как следствие отрицательного эффекта от неправильной укладки белка, которая, помимо вызываемых ею энергетических издержек, может быть еще и токсичной для клетки (Drummond et al., 2005; Drummond and Wilke, 2008). Не углубляясь в детали, заметим, что эта интуитивно привлекательная модель может естественным образом объяснить отрицательную корреляцию между экспрессией генов и эволюцией генных последовательностей: очевидно, что негативный эффект от неправильной укладки должен быть выше для высокоэкспрессированных белков, чем для белков, производимых в небольших количествах. Другими словами, уровень экспрессии – это линза, которая увеличивает любое негативное влияние на приспособленность, связанное с данной последовательностью белков, и важнейшее из таких влияний обусловлено неправильной укладкой белка. Таким образом, гены высокоэкспрессируемых белков подвергаются бо́льшим ограничениям, следствием чего является низкая скорость их эволюции. Эта гипотеза совместима с твердо установленным принципом предпочтительного выбора кодона (среди синонимичных кодонов чаще встречается оптимальный)[37] в высокоэкспрессируемых и высококонсервативных белок-кодирующих генах, а также с положительной корреляцией между Ka и Ks. Согласно гипотезе эволюции, движимой ошибками укладки, эволюция синонимичных сайтов ограничена, по крайней мере частично, теми же самыми факторами, что и эволюция последовательности белка, поскольку выбор оптимального кодона, обеспечивающий более быструю и точную трансляцию, особенно важен для высокоэкспрессированных белков и для тех конкретных позиций, которые влияют на укладку белка. Таким же образом можно объяснить и эволюцию 3-НТУ – этот нетранслируемый участок используется для регуляции процесса трансляции.
В начале третьего тысячелетия геномика и системная биология полностью преобразили область эволюционных исследований. Доступность множества данных по геномным последовательностям позволила проанализировать и сравнить распределения скоростей эволюции для полных наборов ортологичных генов в различных таксонах, а также изучить взаимосвязи скоростей эволюции ортологов в различных эволюционных линиях. Значения скоростей эволюции по несинонимичным сайтам в ортологичных генах могут различаться на три-четыре порядка, и это распределение значений гораздо шире, чем распределение скоростей по синонимичным сайтам. Замечательно, что формы графиков распределений по ортологичным белкам исключительно похожи, практически одинаковы для всех изученных клеточных форм жизни, от бактерий и архей до млекопитающих (см. рис. 4–2; Grishin et al., 2000; Wolf et al., 2009). Все эти распределения имеют так называемую логарифмически нормальную форму, то есть распределение логарифма эволюционной скорости близко к нормальному (распределению Гаусса, функция плотности вероятности которого имеет колоколообразную форму). В теории случайных процессов такая форма обычно представляет собой результат произведения многих независимых случайных величин. Универсальность функции распределения среди различных организмов, обладающих глубокими различиями в функциональной организации и сильно различающихся по размеру геномов, представляется неожиданной и может указывать на существование фундаментальных, простых объяснений, которые мы и обсудим в этой главе.
Прогресс в системной биологии позволил измерить корреляции между скоростью эволюции и всеми возможными молекулярно-фенотипическими величинами, такими как уровень экспрессии, концентрации белков, белок-белковые взаимодействия, фенотипический эффект генной мутации и другими (Koonin and Wolf, 2006). Эти поиски корреляций стали практически самостоятельной областью исследований, цель которых, однако, состоит не в описании самих корреляций, а в построении физически осмысленной модели эволюции геномов и феномов. Было найдено много важных корреляций, что позволило увидеть существование некоторых закономерностей, несмотря на «зашумленность» молекулярно-фенотипических данных (особенно данных, полученных на ранних этапах исследований). На рис. 4–3 представлена простая и наглядная, хоть и неизбежно упрощенная общая картина результатов исследований (Wolf et al., 2006). Обобщение результатов показывает, что существуют два обширных класса переменных:
1. Интенсивные, эволюционные переменные – различные скорости геномных изменений, включая эволюцию последовательностей, потерю гена, перестройку генома и другие виды эволюционных процессов.
2. Экстенсивные, фенотипические переменные – скорость экспрессии, скорость трансляции, концентрация белка, частота взаимодействия с другими изучаемыми объектами.
Корреляции внутри каждого из двух классов обычно положительные, а корреляции между двумя классами – отрицательные (рис. 4–3). Эта закономерность предполагает модель «статуса генов», в которой высокостатусные гены эволюционируют медленно, имеют высокий уровень экспрессии и взаимодействуют со многими другими генами. Гены с низким статусом меняются быстро и имеют низкий уровень экспрессии и меньшее число партнеров (рис. 4–4).
Сильнейшая, универсальная связь между эволюционными и молекулярно-фенотипическими переменными состоит в отрицательной корреляции скорости эволюции белок-кодирующих генов и уровня экспрессии: высокоэкспрессированные гены эволюционируют медленно. Эта зависимость наблюдается у всех организмов, для которых есть данные по экспрессии генов (Drummond et al., 2006; Drummond and Wilke, 2008; Pal et al., 2001). Поскольку, как отмечено выше, существует положительная корреляция между Ка и Ks, неудивительно, что скорости эволюции синонимических и несинонимических сайтов связаны с уровнем экспрессии гена качественно одним и тем же образом. Более неожиданно то, что зависимость между экспрессией и скоростью эволюции соблюдается и для 3’-нетранслируемого участка (НТУ), хотя и не обнаружена для 5’-НТУ (Jordan et al., 2004). Эта универсальная отрицательная корреляция проявляется еще сильнее, если сравнивать скорость эволюции напрямую с экспериментально измеренными концентрациями белка (Schrimpf et al., 2009).
Открытие универсальной связи между экспрессией генов и их эволюцией стимулировало смелую попытку новой интерпретации, согласно которой эволюция белков определяется в большей степени принципами структуры и укладки белка, общими для всех организмов, чем его уникальными биологическими функциями. Было выдвинуто предположение, впервые – в работе Алана Драмонда и Клауса Вилке, о том, что главным фактором отбора в эволюции белка является его устойчивость к неправильной укладке. Согласно этой гипотезе, влияние мутации, как геномной, так и фенотипической (вызванной ошибками трансляции), на приспособленность организма в первую очередь рассматривается как следствие отрицательного эффекта от неправильной укладки белка, которая, помимо вызываемых ею энергетических издержек, может быть еще и токсичной для клетки (Drummond et al., 2005; Drummond and Wilke, 2008). Не углубляясь в детали, заметим, что эта интуитивно привлекательная модель может естественным образом объяснить отрицательную корреляцию между экспрессией генов и эволюцией генных последовательностей: очевидно, что негативный эффект от неправильной укладки должен быть выше для высокоэкспрессированных белков, чем для белков, производимых в небольших количествах. Другими словами, уровень экспрессии – это линза, которая увеличивает любое негативное влияние на приспособленность, связанное с данной последовательностью белков, и важнейшее из таких влияний обусловлено неправильной укладкой белка. Таким образом, гены высокоэкспрессируемых белков подвергаются бо́льшим ограничениям, следствием чего является низкая скорость их эволюции. Эта гипотеза совместима с твердо установленным принципом предпочтительного выбора кодона (среди синонимичных кодонов чаще встречается оптимальный)[37] в высокоэкспрессируемых и высококонсервативных белок-кодирующих генах, а также с положительной корреляцией между Ka и Ks. Согласно гипотезе эволюции, движимой ошибками укладки, эволюция синонимичных сайтов ограничена, по крайней мере частично, теми же самыми факторами, что и эволюция последовательности белка, поскольку выбор оптимального кодона, обеспечивающий более быструю и точную трансляцию, особенно важен для высокоэкспрессированных белков и для тех конкретных позиций, которые влияют на укладку белка. Таким же образом можно объяснить и эволюцию 3-НТУ – этот нетранслируемый участок используется для регуляции процесса трансляции.