Эндосимбиоз
Гипотеза о том, что некоторые органеллы эукариотических клеток, в частности хлоропласты растений, произошли от бактерий, не намного моложе «Происхождения…» Дарвина: некоторые исследователи высказали эту идею в конце XIX века на основе микроскопического исследования клеток растений, показавшего заметное структурное сходство между хлоропластами и цианобактериями (известными тогда как сине-зеленые водоросли). Концепция симбиогенетической эволюции была последовательно представлена Константином Мережковским в начале XX века[28]. Тем не менее в течение первых двух третей ХХ века гипотеза эндосимбиоза оставалась маргинальным теоретизированием. Такое восприятие изменилось вскоре после появления в 1967 году революционной статьи Линн Саган (Маргулис), где она обобщила данные о сходстве органелл и бактерий, и в особенности о совершенно неожиданно открытых незадолго до того геномах и системах трансляции органелл. Саган сделала вывод, что не только хлоропласты, но и митохондрии произошли от эндосимбиотических бактерий (Sagan, 1967). Последующие исследования, и в особенности филогенетический анализ как генов, содержащихся в митохондриальном геноме, так и генов, кодирующих белки, которые функционируют в митохондриях и, видимо, были перенесены из митохондриального в ядерный геном, превратили гипотезу эндосимбиоза в устоявшуюся концепцию с чрезвычайно прочными эмпирическими основаниями (Lang et al., 1999). Кроме того, эти филогенетические исследования убедительно продемонстрировали происхождение митохондрий от определенной группы бактерий, α-протеобактерий. Фундаментальная роль в эволюции, которая отводится уникальным (или крайне редким) событиям, таким как эндосимбиоз, не совместима ни с градуализмом, ни с униформизмом, и является одной из основных тем в остальной части этой книги, в частности в главах 7 и 12.
Канализация и устойчивость в эволюции
Выдающийся эволюционный генетик Конрад Уоддингтон выдвинул неортодоксальную идею канализации развития, которая является частью его общей концепции эпигенетического ландшафта[29]. Эпигенетический ландшафт – это отображение решений, принимаемых развивающимся эмбрионом, так что развитие происходит за счет движения вдоль долин, по которым проходят группы сходных траекторий. Таким образом, относительно небольшие возмущения, вызванные либо факторами окружающей среды, либо мутациями, не влияют на развитие, то есть биологические системы существенно устойчивы. Согласно концепции Уоддингтона, эта устойчивость является эволюционировавшим, адаптивным свойством биологических систем. Внешнее давление может нарушить канализацию и обнаружить скрытую изменчивость, увеличивая тем самым эволюционный потенциал популяции (Waddington and Robertson, 1966). Во времена Уоддингтона эти идеи были за пределами главного русла эволюционной биологии, но в новой концепции эволюции надежность и эволюционный потенциал занимают центральное место, как обсуждается в главе 9.
Краткий обзор и перспектива
Вскоре после того, как была создана СТЭ, в эволюционной биологии произошли разительные перемены: эволюцию стало возможно проследить непосредственно к ее основе, эволюционирующему геному. На самом глубоком концептуальном уровне эволюция путем естественного отбора и дрейфа является неизбежным следствием подверженной ошибкам репликации генетической информации, кодируемой по цифровому принципу. Эволюция перестала быть несколько абстрактным процессом накопления мутаций, наблюдаемых лишь косвенно через их фенотипический эффект. Напротив, эволюция в настоящее время рассматривается как накопление конкретных изменений различного рода, больших и малых, выявляемых прямым сравнением все более доступных генных и геномных последовательностей. Наличие градиента дивергенции последовательностей от близкородственных к далеким видам само по себе является лучшим доказательством эволюции. Эта тенденция воплощается в теории (почти) нейтральной молекулярной эволюции и, на более практическом уровне, позволяет строить осмысленные филогенетические деревья. Молекулярная филогенетика достигла высшей точки с построением трехдоменного древа жизни, первоначально обнаруженного через филогении рРНК, а затем поддержанного филогениями многих белков. Анализ древних паралогов поместил корень на бактериальную ветвь трехдоменного ДЖ. Тем не менее первые выявленные расхождения между топологиями деревьев отдельных генов подсказали, что дерево рРНК не сможет рассказать всей истории эволюции жизни.
Сравнение первых секвенированных геномных последовательностей небольших вирусов положило начало эволюционной геномике. Стало ясно, что с помощью сравнительного анализа могут быть построены структурные и функциональные карты геномов, которые нельзя было охарактеризовать никаким иным способом, и что поразительная консервативность ключевых генов идет рука об руку с пластичностью архитектуры генома.
Одновременно с завершением развития СТЭ на заре молекулярной эволюции и молекулярной филогенетики эволюционная биология догеномной эпохи включала несколько концепций, таких как пандативы и канализация, выходящих за рамки неодарвинизма. В результате быстрый расцвет геномики в 1990-х годах происходил на фоне сложного, разнообразного ландшафта эволюционной теории и методологии.
Сравнение первых секвенированных геномных последовательностей небольших вирусов положило начало эволюционной геномике. Стало ясно, что с помощью сравнительного анализа могут быть построены структурные и функциональные карты геномов, которые нельзя было охарактеризовать никаким иным способом, и что поразительная консервативность ключевых генов идет рука об руку с пластичностью архитектуры генома.
Одновременно с завершением развития СТЭ на заре молекулярной эволюции и молекулярной филогенетики эволюционная биология догеномной эпохи включала несколько концепций, таких как пандативы и канализация, выходящих за рамки неодарвинизма. В результате быстрый расцвет геномики в 1990-х годах происходил на фоне сложного, разнообразного ландшафта эволюционной теории и методологии.
Рекомендуемая дополнительная литература
Dawkins, Richard. (1976/2006) The Selfish Gene. Oxford: Oxford University Press. (Перевод: Докинз Р. Эгоистичный ген / Пер. с англ. Н. Фоминой. М.: Мир, 1993.)
Книга неоценимой концептуальной значимости, в которой впервые представлена плодотворная идея отбора на уровне индивидуальных генов.
Gould, S. J., and N. Eldredge. (1993) Punctuated Equilibrium Comes of Age. Nature 366: 223–227.
Краткий, но убедительный обзор данных в поддержку модели прерывистого равновесия.
Gould, S. J., and R. C. Lewontin. (1979). The Spandrels of San Marco and the Panglossian Paradigm: A Critique of the Adaptationist Programme. Proceedings of the Royal Society of London B Biological Sciences 205: 581–598.
Вероятно, одна из самых выдающихся статей во всей истории биологии, замечательная как непримиримым отстаиванием плюралистического взгляда на эволюцию в противовес панадаптационизму, так и духом Возрождения, которым пропитан весь текст.
Jacob, F. (1977) Evolution and Tinkering. Science 196: 1,161—1,166.
Великолепно написанная статья, которая остается незаменимой как опровержение идеи «естественный отбор как дизайнер» и обоснование определяющей роли исторического стечения обстоятельств в эволюции.
Kimura, Motoo. (1983) The Neutral Theory of Molecular Evolution. Cambridge, MA: Cambridge University Press. (Перевод: Кимура М. Молекулярная эволюция: теория нейтральности Пер. с англ. М.: Мир, 1985.)
Дефинитивное изложение теории нейтральной эволюции ее создателем, сочетающее скрупулезный популяционно-генетический анализ с совершенно прозрачными пояснениями для биологов. Несмотря на то что практические аспекты книги несколько устарели, она остается непревзойденной по ясности изложения и легко читаемой.
Koonin, E. V., and V. V. Dolja. (1993) Evolution and Taxonomy of Positive-Strand RNA Viruses: Implications of Comparative Analysis of Amino Acid Sequences. Critical Reviews in Biochemistry and Molecular Biology 28: 375–430.
Исчерпывающий обзор ранних результатов сравнительной и эволюционной геномики обширного класса малых РНК-вирусов, одна из первых попыток глубокой эволюционной реконструкции. Основные выводы сохраняют свое значение, несмотря на последующий рост количества разнообразных секвенированных вирусных геномов.
Nei, Masatoshi, and Sudhir Kumar. (2000) Molecular Evolution and Phylogenetics. Oxford: Oxford University Press. (Перевод: Ней М., Кумар С. Молекулярная эволюция и филогенетика / Пер. с англ. Киев: КВЩ, 2004.)
Техническое, но вполне доступное описание широко используемых филогенетических методов, написанное автором метода объединения ближайших соседей и одним из авторов широко используемого филогенетического пакета MEGA.
Nielsen, R. (2009) Adaptionism —30 Years After Gould and Lewontin. Evolution 63: 2,487—2,490.
Взгляд на «Пандативы Святого Марка» 30 лет спустя.
Ohno, Susumu. (1970) Evolution by Gene Duplication. Vienna: Springer.
Классическая книга, в которой описывается концепция дупликации генов как центральный путь развития эволюции.
Ohta, T., and J. H. Gillespie. (1996) Development of Neutral and Nearly Neutral Theories. Theoretical Population Biology 49: 128–142.
Историческое и концептуальное освещение нейтральной и почти нейтральной теорий двумя выдающимися исследователями молекулярной эволюции.
Woese, C. R. (1987) Bacterial Evolution. Microbiology Reviews 51: 221–271.
Блистательная, исчерпывающая работа, обобщающая ранние результаты филогенетики 16S-рРНК.
Woese, C. R., and N. Goldenfeld. (2009) How the Microbial World Saved Evolution from the Scylla of Molecular Biology and the Charybdis of the Modern Synthesis. Microbiology and Molecular Biology Reviews 73: 14–21.
Эссе к 200-летию Дарвина, подчеркивающее исключительную важность геномики микроорганизмов для нового понимания эволюции.
Книга неоценимой концептуальной значимости, в которой впервые представлена плодотворная идея отбора на уровне индивидуальных генов.
Gould, S. J., and N. Eldredge. (1993) Punctuated Equilibrium Comes of Age. Nature 366: 223–227.
Краткий, но убедительный обзор данных в поддержку модели прерывистого равновесия.
Gould, S. J., and R. C. Lewontin. (1979). The Spandrels of San Marco and the Panglossian Paradigm: A Critique of the Adaptationist Programme. Proceedings of the Royal Society of London B Biological Sciences 205: 581–598.
Вероятно, одна из самых выдающихся статей во всей истории биологии, замечательная как непримиримым отстаиванием плюралистического взгляда на эволюцию в противовес панадаптационизму, так и духом Возрождения, которым пропитан весь текст.
Jacob, F. (1977) Evolution and Tinkering. Science 196: 1,161—1,166.
Великолепно написанная статья, которая остается незаменимой как опровержение идеи «естественный отбор как дизайнер» и обоснование определяющей роли исторического стечения обстоятельств в эволюции.
Kimura, Motoo. (1983) The Neutral Theory of Molecular Evolution. Cambridge, MA: Cambridge University Press. (Перевод: Кимура М. Молекулярная эволюция: теория нейтральности Пер. с англ. М.: Мир, 1985.)
Дефинитивное изложение теории нейтральной эволюции ее создателем, сочетающее скрупулезный популяционно-генетический анализ с совершенно прозрачными пояснениями для биологов. Несмотря на то что практические аспекты книги несколько устарели, она остается непревзойденной по ясности изложения и легко читаемой.
Koonin, E. V., and V. V. Dolja. (1993) Evolution and Taxonomy of Positive-Strand RNA Viruses: Implications of Comparative Analysis of Amino Acid Sequences. Critical Reviews in Biochemistry and Molecular Biology 28: 375–430.
Исчерпывающий обзор ранних результатов сравнительной и эволюционной геномики обширного класса малых РНК-вирусов, одна из первых попыток глубокой эволюционной реконструкции. Основные выводы сохраняют свое значение, несмотря на последующий рост количества разнообразных секвенированных вирусных геномов.
Nei, Masatoshi, and Sudhir Kumar. (2000) Molecular Evolution and Phylogenetics. Oxford: Oxford University Press. (Перевод: Ней М., Кумар С. Молекулярная эволюция и филогенетика / Пер. с англ. Киев: КВЩ, 2004.)
Техническое, но вполне доступное описание широко используемых филогенетических методов, написанное автором метода объединения ближайших соседей и одним из авторов широко используемого филогенетического пакета MEGA.
Nielsen, R. (2009) Adaptionism —30 Years After Gould and Lewontin. Evolution 63: 2,487—2,490.
Взгляд на «Пандативы Святого Марка» 30 лет спустя.
Ohno, Susumu. (1970) Evolution by Gene Duplication. Vienna: Springer.
Классическая книга, в которой описывается концепция дупликации генов как центральный путь развития эволюции.
Ohta, T., and J. H. Gillespie. (1996) Development of Neutral and Nearly Neutral Theories. Theoretical Population Biology 49: 128–142.
Историческое и концептуальное освещение нейтральной и почти нейтральной теорий двумя выдающимися исследователями молекулярной эволюции.
Woese, C. R. (1987) Bacterial Evolution. Microbiology Reviews 51: 221–271.
Блистательная, исчерпывающая работа, обобщающая ранние результаты филогенетики 16S-рРНК.
Woese, C. R., and N. Goldenfeld. (2009) How the Microbial World Saved Evolution from the Scylla of Molecular Biology and the Charybdis of the Modern Synthesis. Microbiology and Molecular Biology Reviews 73: 14–21.
Эссе к 200-летию Дарвина, подчеркивающее исключительную важность геномики микроорганизмов для нового понимания эволюции.
Глава 3. Сравнительная геномика: эволюционирующие геномные ландшафты
Важность перехода к геномике
В догеномную эру были установлены фундаментальные принципы молекулярной эволюции и было сделано немало конкретных наблюдений, имеющих большое значение и повлиявших на основы эволюционной биологии (см. гл. 1 и 2). Но масштабные работы по расшифровке геномов, начавшиеся в середине 90-х и стремительно развивавшиеся в новом тысячелетии, качественно изменили всю эволюционную биологию. Важность обширной базы данных геномных последовательностей, имеющих различную степень расхождения, очевидна. Этот материал дает исследователям возможность изучать механизмы и отдельные события эволюции с необходимой статистической точностью и выявлять даже самые малозаметные эволюционные подвижки. Как бы то ни было, в эволюционной биологии получение разнообразных и полных геномных последовательностей чрезвычайно важно далеко не только ради накопления количества данных. Действительно, лишь полностью расшифрованный геном (в отличие от, скажем, расшифрованного лишь на 95 процентов) дает исследователю объективное и непредвзятое представление о генном репертуаре той или иной формы жизни. Иными словами, исследователь может определить присутствие в организме тех или иных генов и, что одинаково важно, их отсутствие. Таким образом, сравнение полных геномов представляет собой единственный удовлетворяющий исследователя путь к реконструкции эволюции. Открывающаяся картина во многом отличается от всего, что можно было себе представить, оставаясь в рамках традиционной эволюционной биологии.
Если мы действительно стремимся «понять» эволюцию, принципиально важно исследовать геномные образцы как вглубь (для этого необходимы геномные последовательности множества близкородственных представителей одного и того же таксона), так и вширь (для этой цели нужны последовательности как можно большего числа различных таксонов – в идеале всех таксонов). Ко времени написания этих строк, в последние дни 2010 года, собрание секвенированных геномов состояло из нескольких тысяч геномов вирусов, более чем тысячи геномов бактерий и архей, а также приблизительно сотни геномов эукариот. Ко времени издания этой книги геномная база данных почти удвоится, а благодаря новому поколению методов секвенирования в предстоящие годы ее темпы роста должны еще более ускориться[30]. Несмотря на то что не все основные таксоны должным образом охвачены, быстро пополняющееся собрание геномов все более отвечает потребностям исследований как в области микроэволюции, так и в области макроэволюции.
Успехи традиционной геномики дополняют и стремительно накапливающиеся в последнее время, обширные по объему данные по метагеномике – а именно всеобъемлющее (или, по меньшей мере, обширное) секвенирование нуклеиновых кислот форм жизни из разнообразных сред обитания. Хотя применяемые в настоящее время в метагеномике подходы обычно не обеспечивают полную расшифровку геномов, они предоставляют бесценную, объективную информацию о разнообразии жизни в различных средах.
В данной главе представлен обзор разнообразия и основных характеристик геномов. В последующих главах подробно исследуется влияние результатов сравнительных геномных исследований на развитие «постсовременной» синтетической теории эволюционной биологии.
Если мы действительно стремимся «понять» эволюцию, принципиально важно исследовать геномные образцы как вглубь (для этого необходимы геномные последовательности множества близкородственных представителей одного и того же таксона), так и вширь (для этой цели нужны последовательности как можно большего числа различных таксонов – в идеале всех таксонов). Ко времени написания этих строк, в последние дни 2010 года, собрание секвенированных геномов состояло из нескольких тысяч геномов вирусов, более чем тысячи геномов бактерий и архей, а также приблизительно сотни геномов эукариот. Ко времени издания этой книги геномная база данных почти удвоится, а благодаря новому поколению методов секвенирования в предстоящие годы ее темпы роста должны еще более ускориться[30]. Несмотря на то что не все основные таксоны должным образом охвачены, быстро пополняющееся собрание геномов все более отвечает потребностям исследований как в области микроэволюции, так и в области макроэволюции.
Успехи традиционной геномики дополняют и стремительно накапливающиеся в последнее время, обширные по объему данные по метагеномике – а именно всеобъемлющее (или, по меньшей мере, обширное) секвенирование нуклеиновых кислот форм жизни из разнообразных сред обитания. Хотя применяемые в настоящее время в метагеномике подходы обычно не обеспечивают полную расшифровку геномов, они предоставляют бесценную, объективную информацию о разнообразии жизни в различных средах.
В данной главе представлен обзор разнообразия и основных характеристик геномов. В последующих главах подробно исследуется влияние результатов сравнительных геномных исследований на развитие «постсовременной» синтетической теории эволюционной биологии.
Эволюция геномных ландшафтов
Поразительное разнообразие геномов
Геном стал первым термином с окончанием «-ом» – и до сих пор является наиболее употребительным термином этой группы[31]. Как это всегда бывает в биологии, определить, что же такое геном, нелегко. Говоря просто, геном – это генетическая информация конкретного организма во всей ее полноте. Существование стабильного ядра унаследованной генетической информации (а более конкретно, генов) вытекает из самого факта существования надежной наследственности, а в терминах более фундаментальных – из принципа подверженной ошибкам репликации (ПОР, см. гл. 2). Однако связь между «генетической информацией во всей ее полноте» и «стабильным ядром» не так уж проста. Стоит, к примеру, задать на первый взгляд невинный вопрос: «Что есть геном кишечной палочки Escherichia coli?» – как тут же возникает целый ряд серьезных затруднений. А вопрос «Что такое геном человека?» вызывает свои, не менее сложные проблемы. Вернемся мы к этому обсуждению позднее (см. гл. 5), а сейчас рассмотрим многообразие геномов, расшифрованных за последние 15 лет.
Новая эра геномики наступила на исходе лета 1995 года. Тогда лаборатория Дж. Крейга Вентера опубликовала результаты секвенирования генома условно-патогенной бактерии гемофильного гриппа Haemophilus influenzae (Fleischmann et al., 1995). В процессе расшифровки геномной последовательности H. influenzae Вентер, Гамильтон Смит и их коллеги усовершенствовали так называемый «метод дробовика». Этот подход грубого деления генома на короткие произвольные участки с расшифровкой их по частям и последующим восстановлением полной геномной последовательности быстро превратил секвенирование длинных нуклеотидных цепочек в рутинное дело. В течение года были расшифрованы геномы некоторых других бактерий, первый геном археи (Methanocaldococcus jannaschii) и первый геном эукариота (пекарские дрожжи Saccharomyces cerevisiae) (Koonin et al., 1996). К 1999 году установился стабильный экспоненциальный рост коллекции секвенированных геномов (см. рис. 3–1).
В диапазоне от вирусов до животных геномы различаются по размеру на шесть порядков – от нескольких тысяч до нескольких миллиардов нуклеотидов; для клеточных организмов, исключая вирусы, ширина диапазона составляет четыре порядка (см. рис. 3–2). По количеству генов диапазон значительно уже и составляет всего около четырех порядков, от двух-трех генов у простейших вирусов до приблизительно 40 тысяч генов у некоторых животных. Если же исключить вирусы и паразитические (симбиотические) бактерии, диапазон по числу генов становится довольно узким, немногим более одного порядка (см. рис. 3–2; Koonin, 2009a; Lynch, 2007c). Кажется весьма удивительным, что млекопитающие или цветковые растения имеют всего примерно в десять раз больше (легко идентифицируемых) генов, чем какая-нибудь средняя свободно живущая бактерия, и лишь примерно в два раза больше, чем бактерия из разряда наиболее сложных (см. рис. 3–2). Далее в книге рассматриваются всевозможные объяснения этих явных ограничений по числу генов в геномах всех форм жизни (см. гл. 5, 7 и 10).
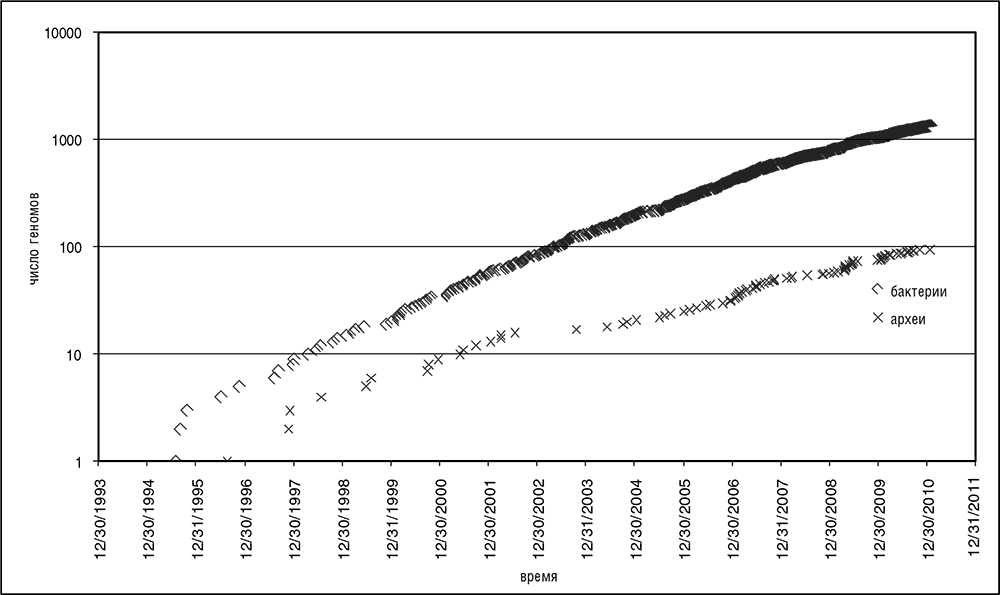 Рис. 3–1. Экспоненциальный рост коллекции секвенированных геномов. Данные с веб-сайта Национального центра биотехнологической информации (www.ncbi.nlm.nih.gov/genome/)
Рис. 3–1. Экспоненциальный рост коллекции секвенированных геномов. Данные с веб-сайта Национального центра биотехнологической информации (www.ncbi.nlm.nih.gov/genome/)
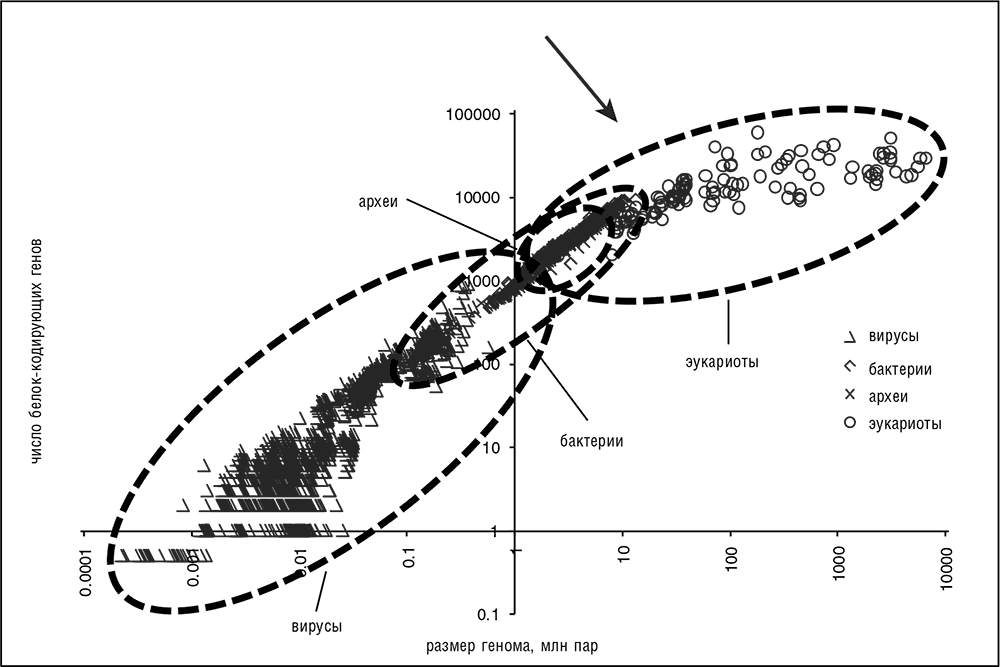 Рис. 3–2. Общий размер геномов и число генов у вирусов, бактерий, архей и эукариот. Данные с веб-сайта Национального центра биотехнологической информации. Представлено в двойном логарифмическом масштабе. Стрелка указывает на точку изменения наклона кривой, соответствующую переходу от «малых» к «большим» геномам.
Рис. 3–2. Общий размер геномов и число генов у вирусов, бактерий, архей и эукариот. Данные с веб-сайта Национального центра биотехнологической информации. Представлено в двойном логарифмическом масштабе. Стрелка указывает на точку изменения наклона кривой, соответствующую переходу от «малых» к «большим» геномам.
Грубо говоря, геномы могут быть разделены на два четко выделенных класса (Koonin, 2009а). Граница, разделяющая эти классы, находится в точке изменения наклона кривой на графике, представленном на рис. 3–2.
1. Геномы со строгим соответствием между размером генома и числом генов. К ним относятся геномы всех вирусов и прокариот, имеющие огромную плотность генов от 0,5 до 2 генов на тысячу пар оснований и очень короткие участки между генами (10–15 процентов длины генома и даже меньше), состоящие главным образом из регуляторных элементов. Иногда, говоря о таких геномах, вспоминают ковер «от стены к стене» (wall to wall genomes)[32], так как они почти полностью состоят из легко определяемых генов. Геномы большинства одноклеточных эукариот демонстрируют несколько меньшую зависимость между размером генома и числом генов, чем геномы вирусов и прокариот, тем не менее они могут быть отнесены к этому же классу.
2. Геномы, у которых нет четкой взаимосвязи между размером генома и числом генов, в частности большие геномы многоклеточных и некоторых одноклеточных эукариот. Здесь в лучшем случае наблюдается слабая корреляция между общим размером генома и числом генов. Соответственно, доля генома, занимаемая межгенными участками (а также другими некодирующими последовательностями, такими как интроны), сильно варьирует. В некоторых наиболее сложных геномах, в частности у млекопитающих, основную часть генома составляют именно некодирующие последовательности.
Вариабельность размеров генома и числа генов дополняется разнообразием в других измерениях – например, в физической организации и композиции нуклеотидов. При рассмотрении как вирусной, так и клеточной формы жизни геномы предстают во всевозможных формах нуклеиновых кислот (подробнее см. в гл. 10). Все геномы клеточных организмов состоят из двухцепочечных ДНК, однако количество геномных сегментов (хромосом) и их размеры, форма (кольцевая или линейная), а также плоидность (число наборов) широко разнятся. Азбучная истина гласит, что прокариоты имеют гаплоидные, простые кольцевые хромосомы, в то время как у геномов эукариот, сильно различающихся по плоидности, гены распределены между множеством линейных хромосом. И хотя такие геномные формы, по-видимому, действительно доминируют, на самом деле разнообразие геномов выходит далеко за рамки такого простого дихотомического разделения. В частности, у многих прокариот имеется несколько хромосом, в отдельных случаях – линейных. Более того, вопреки распространенному заблуждению, у прокариот большинство клеток не гаплоидные, то есть они содержат несколько копий генома.
Новая эра геномики наступила на исходе лета 1995 года. Тогда лаборатория Дж. Крейга Вентера опубликовала результаты секвенирования генома условно-патогенной бактерии гемофильного гриппа Haemophilus influenzae (Fleischmann et al., 1995). В процессе расшифровки геномной последовательности H. influenzae Вентер, Гамильтон Смит и их коллеги усовершенствовали так называемый «метод дробовика». Этот подход грубого деления генома на короткие произвольные участки с расшифровкой их по частям и последующим восстановлением полной геномной последовательности быстро превратил секвенирование длинных нуклеотидных цепочек в рутинное дело. В течение года были расшифрованы геномы некоторых других бактерий, первый геном археи (Methanocaldococcus jannaschii) и первый геном эукариота (пекарские дрожжи Saccharomyces cerevisiae) (Koonin et al., 1996). К 1999 году установился стабильный экспоненциальный рост коллекции секвенированных геномов (см. рис. 3–1).
В диапазоне от вирусов до животных геномы различаются по размеру на шесть порядков – от нескольких тысяч до нескольких миллиардов нуклеотидов; для клеточных организмов, исключая вирусы, ширина диапазона составляет четыре порядка (см. рис. 3–2). По количеству генов диапазон значительно уже и составляет всего около четырех порядков, от двух-трех генов у простейших вирусов до приблизительно 40 тысяч генов у некоторых животных. Если же исключить вирусы и паразитические (симбиотические) бактерии, диапазон по числу генов становится довольно узким, немногим более одного порядка (см. рис. 3–2; Koonin, 2009a; Lynch, 2007c). Кажется весьма удивительным, что млекопитающие или цветковые растения имеют всего примерно в десять раз больше (легко идентифицируемых) генов, чем какая-нибудь средняя свободно живущая бактерия, и лишь примерно в два раза больше, чем бактерия из разряда наиболее сложных (см. рис. 3–2). Далее в книге рассматриваются всевозможные объяснения этих явных ограничений по числу генов в геномах всех форм жизни (см. гл. 5, 7 и 10).
Грубо говоря, геномы могут быть разделены на два четко выделенных класса (Koonin, 2009а). Граница, разделяющая эти классы, находится в точке изменения наклона кривой на графике, представленном на рис. 3–2.
1. Геномы со строгим соответствием между размером генома и числом генов. К ним относятся геномы всех вирусов и прокариот, имеющие огромную плотность генов от 0,5 до 2 генов на тысячу пар оснований и очень короткие участки между генами (10–15 процентов длины генома и даже меньше), состоящие главным образом из регуляторных элементов. Иногда, говоря о таких геномах, вспоминают ковер «от стены к стене» (wall to wall genomes)[32], так как они почти полностью состоят из легко определяемых генов. Геномы большинства одноклеточных эукариот демонстрируют несколько меньшую зависимость между размером генома и числом генов, чем геномы вирусов и прокариот, тем не менее они могут быть отнесены к этому же классу.
2. Геномы, у которых нет четкой взаимосвязи между размером генома и числом генов, в частности большие геномы многоклеточных и некоторых одноклеточных эукариот. Здесь в лучшем случае наблюдается слабая корреляция между общим размером генома и числом генов. Соответственно, доля генома, занимаемая межгенными участками (а также другими некодирующими последовательностями, такими как интроны), сильно варьирует. В некоторых наиболее сложных геномах, в частности у млекопитающих, основную часть генома составляют именно некодирующие последовательности.
Вариабельность размеров генома и числа генов дополняется разнообразием в других измерениях – например, в физической организации и композиции нуклеотидов. При рассмотрении как вирусной, так и клеточной формы жизни геномы предстают во всевозможных формах нуклеиновых кислот (подробнее см. в гл. 10). Все геномы клеточных организмов состоят из двухцепочечных ДНК, однако количество геномных сегментов (хромосом) и их размеры, форма (кольцевая или линейная), а также плоидность (число наборов) широко разнятся. Азбучная истина гласит, что прокариоты имеют гаплоидные, простые кольцевые хромосомы, в то время как у геномов эукариот, сильно различающихся по плоидности, гены распределены между множеством линейных хромосом. И хотя такие геномные формы, по-видимому, действительно доминируют, на самом деле разнообразие геномов выходит далеко за рамки такого простого дихотомического разделения. В частности, у многих прокариот имеется несколько хромосом, в отдельных случаях – линейных. Более того, вопреки распространенному заблуждению, у прокариот большинство клеток не гаплоидные, то есть они содержат несколько копий генома.
Древние гены составляют в геноме большинство и имеют отчетливую эволюционную судьбу
Как уже упоминалось в предыдущей главе, Эрнст Майр, великий эволюционист XX века и один из основателей СТЭ, с уверенностью предсказывал исходя из больших фенотипических различий между организмами, что гены разных организмов, даже близкородственных, не будут иметь узнаваемого сходства. Ошибочность этого предсказания оказалась просто феерической, что само по себе делает его нетривиальным и ценным. Сравнение последовательностей даже в догеномный период выявило высокий консерватизм последовательностей у некоторых гомологичных белков и молекул некодирующих РНК по всему спектру жизни, от бактерий до млекопитающих (см. предыдущую главу). Более того, высокая степень сходства последовательностей существует у древних паралогов, которые, по-видимому, происходят от копий, ведущих свое происхождение от LUCA (Gogarten et al., 1989; Iwabe et al., 1989). Геномика позволяет перевести это общее понимание в количественное разбиение генов любого генома на классы эволюционной консервативности (см. рис. 3–3; Koonin and Wolf, 2008b).
Ключевое открытие сравнительной геномики состоит в том, что большинство генов в каждом геноме могут считаться высококонсервативными – они имеют легко обнаруживаемые гомологи в организмах, разделяемых сотнями миллионов лет эволюции (например, в случае генов человека, на уровне общего предка позвоночных; см. рис. 3–3; Wolf et al., 2009). Это открытие демонстрирует поразительную устойчивость последовательностей РНК и белков в процессе эволюции: типичное время исчезновения сходства последовательностей у гомологичных генов сравнимо со временем существования жизни на Земле. Помимо основополагающего значения, данный факт имеет огромные практические последствия: благодаря ему, прежде всего, сравнительная геномика становится крайне информативной и действенной.
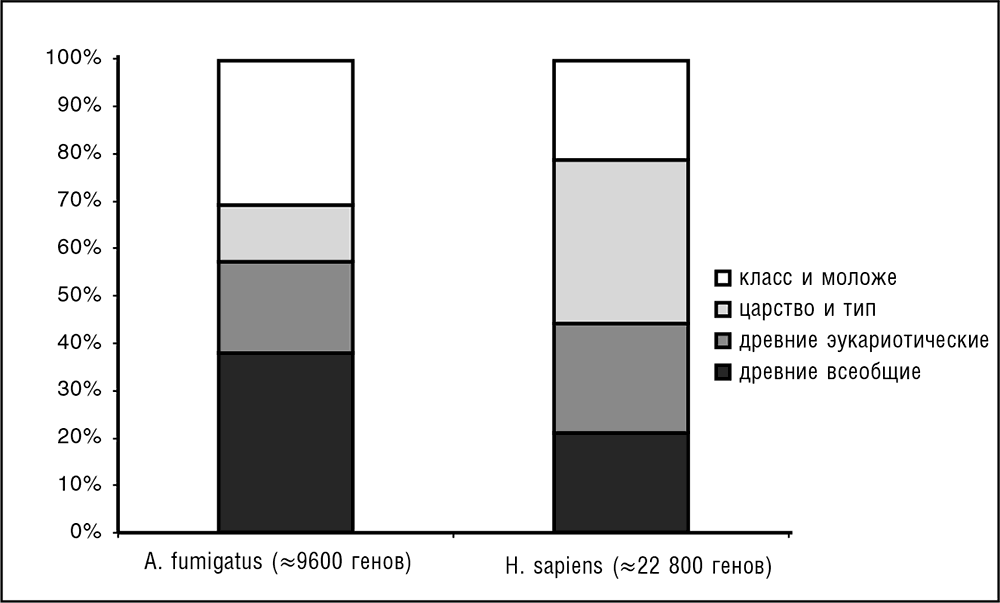 Рис. 3–3. Распределение генов по эволюционному возрасту. «Эволюционный возраст» соответствует самому старому таксономическому узлу, в котором могут быть определены гомологи для белка, производимого данным геном. В частности, для человека древние всеобщие означает «гомологи, обнаруживаемые у прокариот», древние эукариотические означает «гомологи, обнаруживаемые у прокариот вне супергруппы униконтов» (см. гл. 7), царство и тип означает «гомологи, обнаруживаемые у животных вне класса млекопитающих», а класс и моложе означает «вне класса млекопитающих гомологи не обнаружены» (данные по Wolf et al., 2009)
Рис. 3–3. Распределение генов по эволюционному возрасту. «Эволюционный возраст» соответствует самому старому таксономическому узлу, в котором могут быть определены гомологи для белка, производимого данным геном. В частности, для человека древние всеобщие означает «гомологи, обнаруживаемые у прокариот», древние эукариотические означает «гомологи, обнаруживаемые у прокариот вне супергруппы униконтов» (см. гл. 7), царство и тип означает «гомологи, обнаруживаемые у животных вне класса млекопитающих», а класс и моложе означает «вне класса млекопитающих гомологи не обнаружены» (данные по Wolf et al., 2009)
Структуру эволюционного процесса определяют не только консервативные последовательности. На протяжении чрезвычайно длительных эволюционных периодов не просто сохраняется сходство последовательностей РНК и белков, но и гены имеют свойство сохранять свою уникальность. Иными словами, большинство генов развиваются как ортологичные линии, с редкими случаями дупликации (Koonin, 2005). Устойчивость ортологии генов становится очевидной благодаря простой процедуре, широко применяемой в сравнительной геномике и позволяющей эффективно выявлять ортологичные наборы генов. При этом ортологи обнаруживаются как «наилучшие совпадения при двунаправленном сравнении» (bidirectional best hits): все закодированные в геноме белковые последовательности сравниваются со всеми белками, закодированными в другом геноме, a затем процедура повторяется в обратном направлении (Tatusov et al., 1997). Пары генов, дающие наилучшие совпадения (те, которые демонстрируют наибольшее сходство последовательностей) при обоих направлениях сравнения, считаются возможными ортологами; нетрудно применить эту процедуру к нескольким видам путем совмещения треугольников двунаправленных совпадений, имеющих общую сторону (см. табл. 3–1). Примечательно, что такой прямолинейный подход в большинстве случаев хорошо срабатывает: к примеру, порядка 70 процентов генов организмов, разделенных приблизительно 100 миллионами лет эволюции, таких как люди и мыши, легко идентифицируются как ортологи при помощи описанной процедуры (Wolf et al., 2009). Если применить простую модификацию этого алгоритма и включить дупликации генов, характерных для одной линии наследования (дупликации, образовавшиеся после расхождения сравниваемых видов), такой подход позволяет идентифицировать наборы ортологов (известных как кластеры ортологичных генов, КОГ) во многих геномах, в том числе столь удаленных друг от друга, как археи и бактерии – представители двух доменов прокариот (см. гл. 5). Более точные и мощные способы обнаружения ортологов требуют подробного анализа филогенетических деревьев (см. табл. 3–1); впрочем, результаты такого анализа обычно близки к тем, что дают более простые методы, основанные только на сравнении последовательностей. Разумеется, для части генов история дупликаций и потерь настолько сложна, что обнаружить КОГ трудно, поэтому они становятся нечеткими кластерами с неопределенной внутренней структурой. По счастью, этих «трудных» генов в каждом геноме относительно немного.
Ключевое открытие сравнительной геномики состоит в том, что большинство генов в каждом геноме могут считаться высококонсервативными – они имеют легко обнаруживаемые гомологи в организмах, разделяемых сотнями миллионов лет эволюции (например, в случае генов человека, на уровне общего предка позвоночных; см. рис. 3–3; Wolf et al., 2009). Это открытие демонстрирует поразительную устойчивость последовательностей РНК и белков в процессе эволюции: типичное время исчезновения сходства последовательностей у гомологичных генов сравнимо со временем существования жизни на Земле. Помимо основополагающего значения, данный факт имеет огромные практические последствия: благодаря ему, прежде всего, сравнительная геномика становится крайне информативной и действенной.
Структуру эволюционного процесса определяют не только консервативные последовательности. На протяжении чрезвычайно длительных эволюционных периодов не просто сохраняется сходство последовательностей РНК и белков, но и гены имеют свойство сохранять свою уникальность. Иными словами, большинство генов развиваются как ортологичные линии, с редкими случаями дупликации (Koonin, 2005). Устойчивость ортологии генов становится очевидной благодаря простой процедуре, широко применяемой в сравнительной геномике и позволяющей эффективно выявлять ортологичные наборы генов. При этом ортологи обнаруживаются как «наилучшие совпадения при двунаправленном сравнении» (bidirectional best hits): все закодированные в геноме белковые последовательности сравниваются со всеми белками, закодированными в другом геноме, a затем процедура повторяется в обратном направлении (Tatusov et al., 1997). Пары генов, дающие наилучшие совпадения (те, которые демонстрируют наибольшее сходство последовательностей) при обоих направлениях сравнения, считаются возможными ортологами; нетрудно применить эту процедуру к нескольким видам путем совмещения треугольников двунаправленных совпадений, имеющих общую сторону (см. табл. 3–1). Примечательно, что такой прямолинейный подход в большинстве случаев хорошо срабатывает: к примеру, порядка 70 процентов генов организмов, разделенных приблизительно 100 миллионами лет эволюции, таких как люди и мыши, легко идентифицируются как ортологи при помощи описанной процедуры (Wolf et al., 2009). Если применить простую модификацию этого алгоритма и включить дупликации генов, характерных для одной линии наследования (дупликации, образовавшиеся после расхождения сравниваемых видов), такой подход позволяет идентифицировать наборы ортологов (известных как кластеры ортологичных генов, КОГ) во многих геномах, в том числе столь удаленных друг от друга, как археи и бактерии – представители двух доменов прокариот (см. гл. 5). Более точные и мощные способы обнаружения ортологов требуют подробного анализа филогенетических деревьев (см. табл. 3–1); впрочем, результаты такого анализа обычно близки к тем, что дают более простые методы, основанные только на сравнении последовательностей. Разумеется, для части генов история дупликаций и потерь настолько сложна, что обнаружить КОГ трудно, поэтому они становятся нечеткими кластерами с неопределенной внутренней структурой. По счастью, этих «трудных» генов в каждом геноме относительно немного.